

Nlrp3-inflammasome activation in non-myeloid-derived cells aggravates diabetic nephropathy
- PMID: 25075770
- PMCID: PMC4284813
- DOI: 10.1038/ki.2014.271
Nlrp3-inflammasome activation in non-myeloid-derived cells aggravates diabetic nephropathy
Abstract
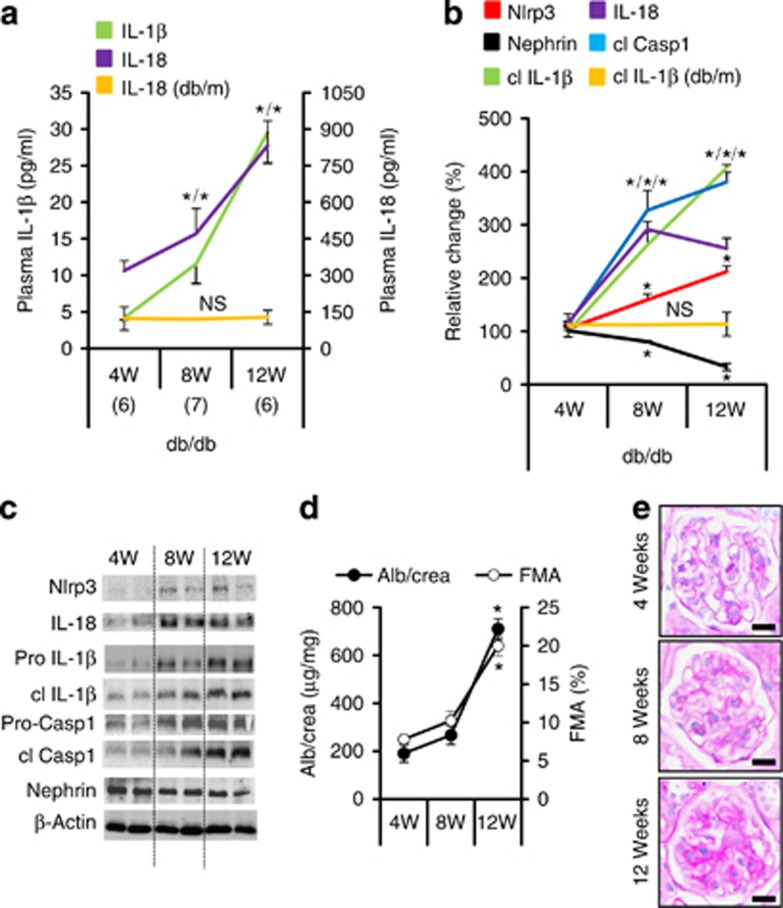
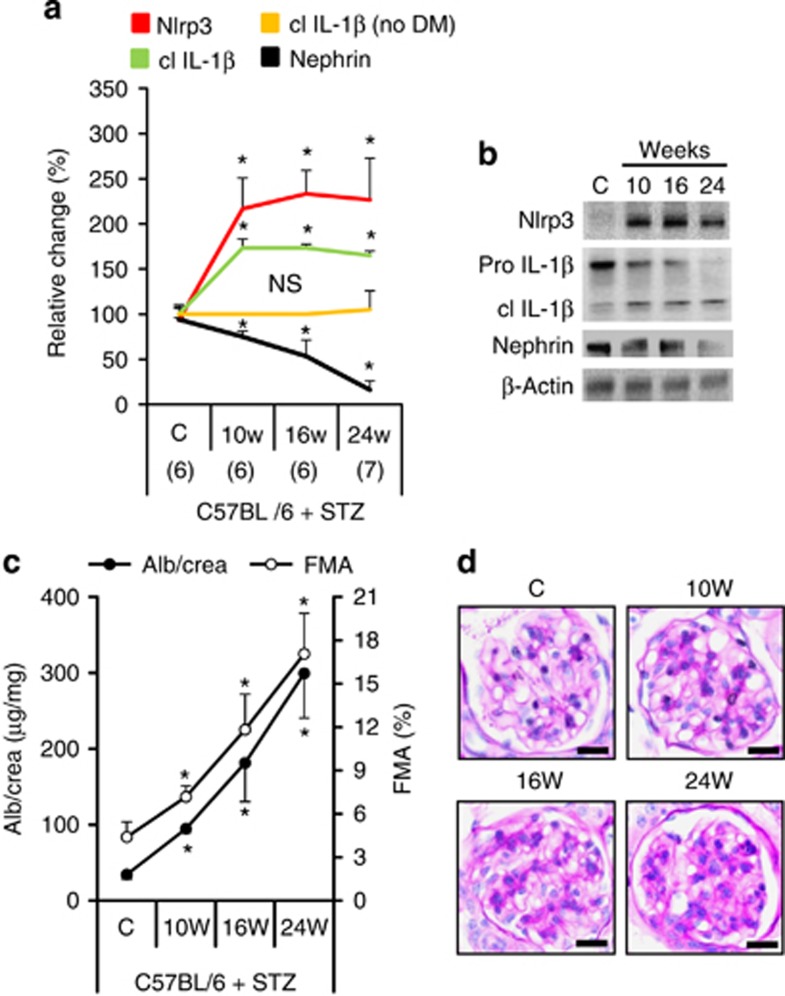
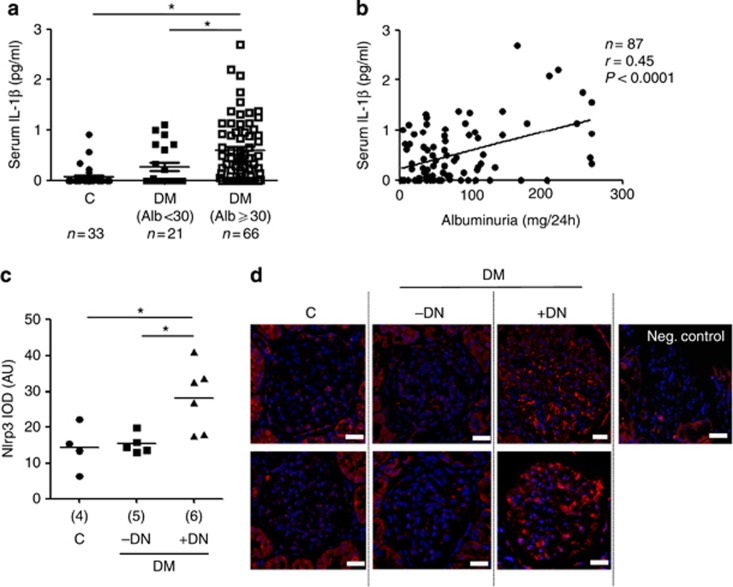
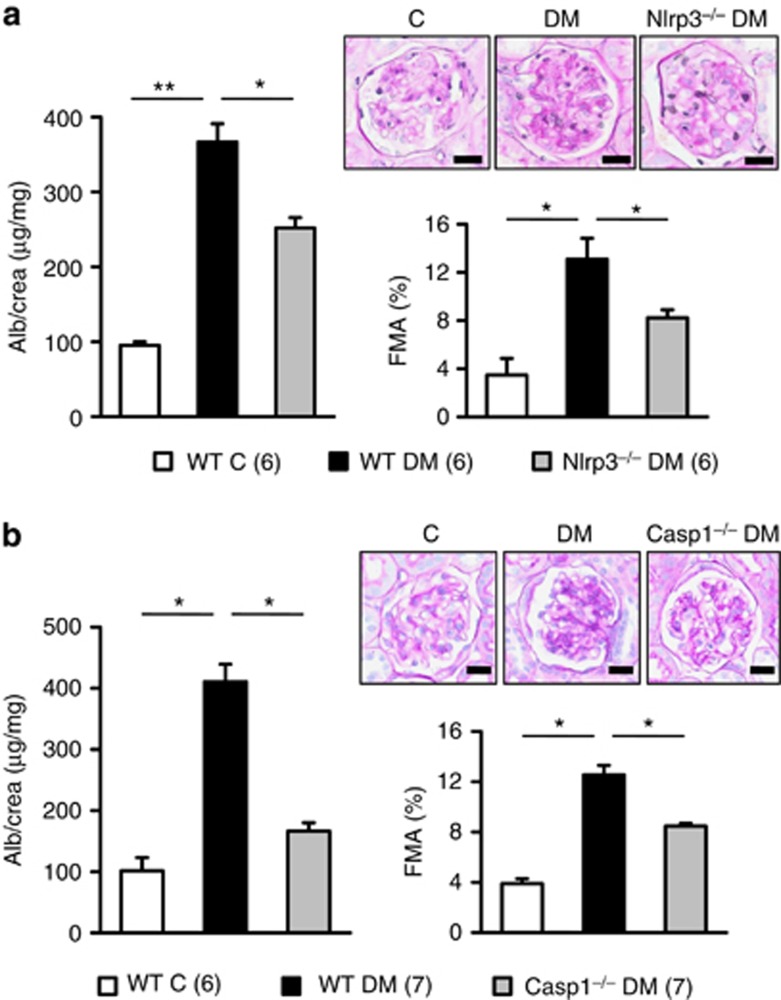
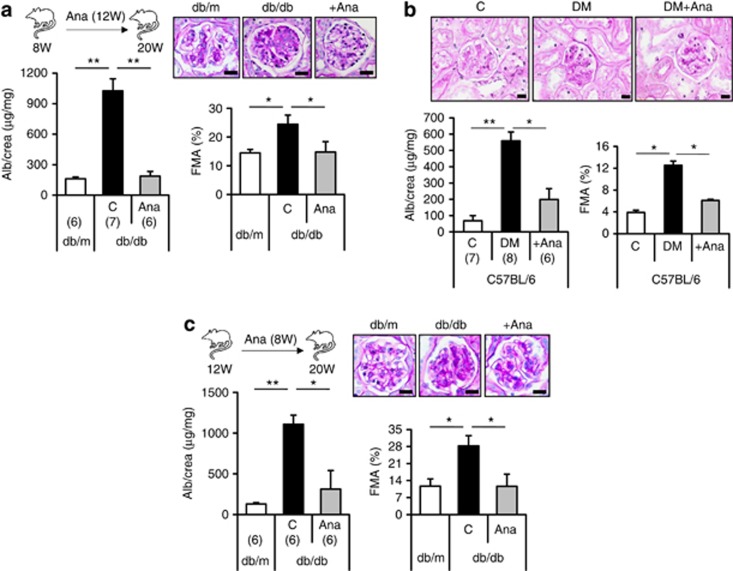
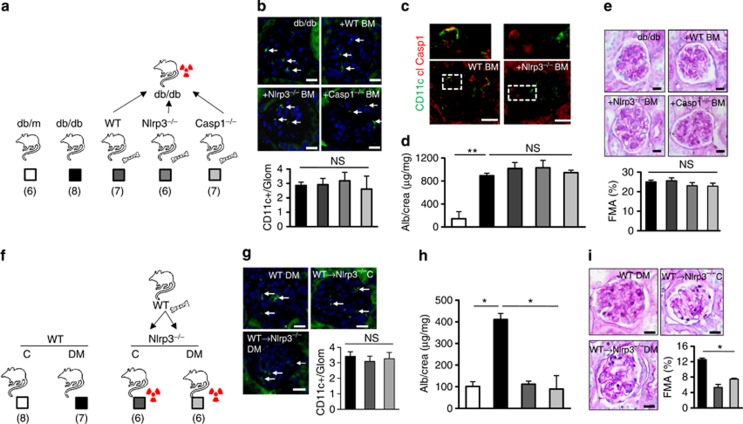
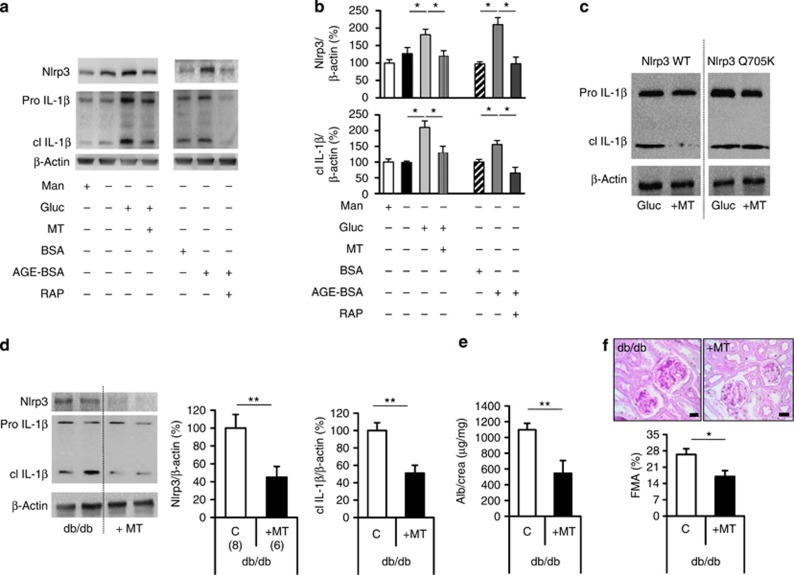
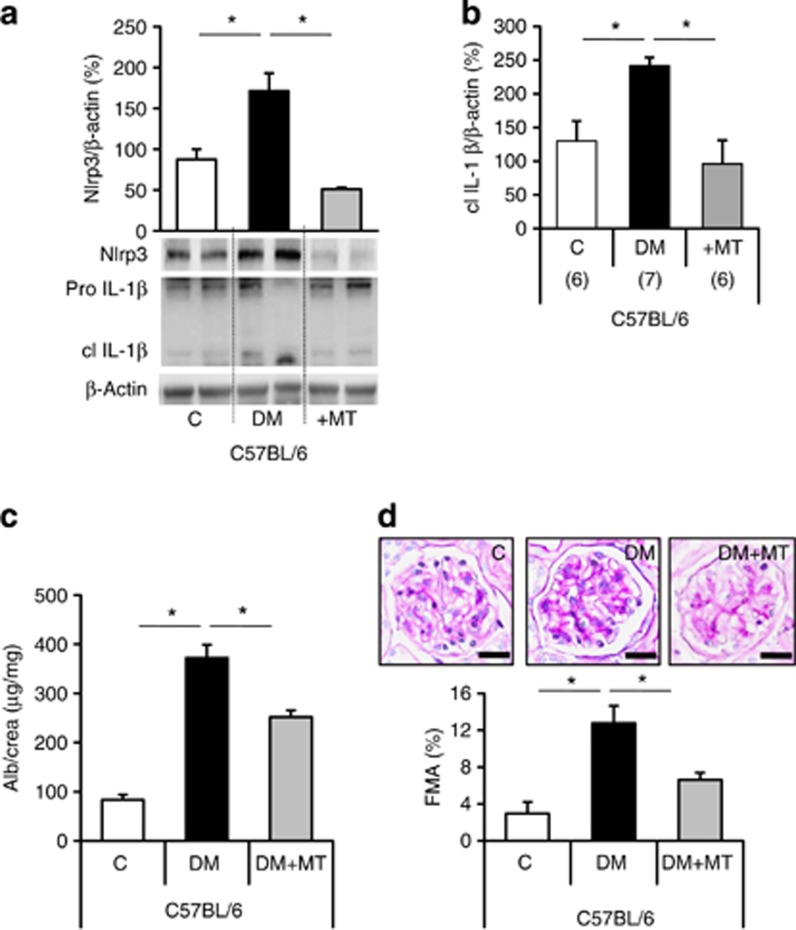
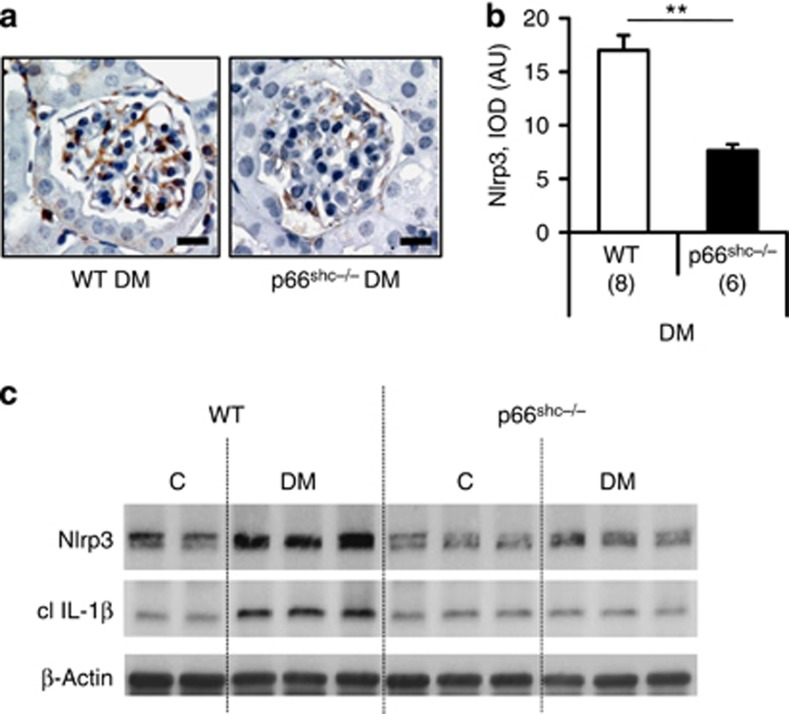
Diabetic nephropathy is a growing health concern with characteristic sterile inflammation. As the underlying mechanisms of this inflammation remain poorly defined, specific therapies targeting sterile inflammation in diabetic nephropathy are lacking. Intriguingly, an association of diabetic nephropathy with inflammasome activation has recently been shown, but the pathophysiological relevance of this finding remains unknown. Within glomeruli, inflammasome activation was detected in endothelial cells and podocytes in diabetic humans and mice and in glucose-stressed glomerular endothelial cells and podocytes in vitro. Abolishing Nlrp3 or caspase-1 expression in bone marrow-derived cells fails to protect mice against diabetic nephropathy. Conversely, Nlrp3-deficient mice are protected against diabetic nephropathy despite transplantation of wild-type bone marrow. Pharmacological IL-1R antagonism prevented or even reversed diabetic nephropathy in mice. Mitochondrial reactive oxygen species (ROS) activate the Nlrp3 inflammasome in glucose or advanced glycation end product stressed podocytes. Inhibition of mitochondrial ROS prevents glomerular inflammasome activation and nephropathy in diabetic mice. Thus, mitochondrial ROS and Nlrp3-inflammasome activation in non-myeloid-derived cells aggravate diabetic nephropathy. Targeting the inflammasome may be a potential therapeutic approach to diabetic nephropathy.
Figures

Comment in
-
Revisiting inflammation in diabetic nephropathy: the role of the Nlrp3 inflammasome in glomerular resident cells.Kidney Int. 2015 Jan;87(1):12-4. doi: 10.1038/ki.2014.322. Kidney Int. 2015. PMID: 25549120
-
Improved renal function in diabetic patients with acute gout treated with anakinra.Kidney Int. 2015 Jul;88(1):195-6. doi: 10.1038/ki.2015.125. Kidney Int. 2015. PMID: 26126095 No abstract available.
-
The Authors Reply.Kidney Int. 2015 Jul;88(1):196-7. doi: 10.1038/ki.2015.128. Kidney Int. 2015. PMID: 26126096 No abstract available.
Similar articles
-
NLRP3 inflammasome-mitochondrion loop in autism spectrum disorder.Free Radic Biol Med. 2024 Nov 20;225:581-594. doi: 10.1016/j.freeradbiomed.2024.10.297. Epub 2024 Oct 19. Free Radic Biol Med. 2024. PMID: 39433111
-
Nerolidol rescues hippocampal injury of diabetic rats through inhibiting NLRP3 inflammasome and regulation of MAPK/AKT pathway.Biofactors. 2024 Nov-Dec;50(6):1076-1100. doi: 10.1002/biof.2058. Epub 2024 Apr 16. Biofactors. 2024. PMID: 38624190
-
Role of NLRP3 Inflammasome in Heart Failure Patients Undergoing Cardiac Surgery as a Potential Determinant of Postoperative Atrial Fibrillation and Remodeling: Is SGLT2 Cotransporter Inhibition an Alternative for Cardioprotection?Antioxidants (Basel). 2024 Nov 14;13(11):1388. doi: 10.3390/antiox13111388. Antioxidants (Basel). 2024. PMID: 39594530 Free PMC article. Review.
-
Breaking the barriers: Overcoming cancer resistance by targeting the NLRP3 inflammasome.Br J Pharmacol. 2025 Jan;182(1):3-25. doi: 10.1111/bph.17352. Epub 2024 Oct 12. Br J Pharmacol. 2025. PMID: 39394867 Review.
-
Depressing time: Waiting, melancholia, and the psychoanalytic practice of care.In: Kirtsoglou E, Simpson B, editors. The Time of Anthropology: Studies of Contemporary Chronopolitics. Abingdon: Routledge; 2020. Chapter 5. In: Kirtsoglou E, Simpson B, editors. The Time of Anthropology: Studies of Contemporary Chronopolitics. Abingdon: Routledge; 2020. Chapter 5. PMID: 36137063 Free Books & Documents. Review.
Cited by
-
Hypoxic human proximal tubular epithelial cells undergo ferroptosis and elicit an NLRP3 inflammasome response in CD1c+ dendritic cells.Cell Death Dis. 2022 Aug 27;13(8):739. doi: 10.1038/s41419-022-05191-z. Cell Death Dis. 2022. PMID: 36030251 Free PMC article.
-
The "ins and outs" of complement-driven immune responses.Immunol Rev. 2016 Nov;274(1):16-32. doi: 10.1111/imr.12472. Immunol Rev. 2016. PMID: 27782335 Free PMC article. Review.
-
Activation of autophagy and nucleotide-binding domain leucine-rich repeat-containing-like receptor family, pyrin domain-containing 3 inflammasome during Leishmania infantum-associated glomerulonephritis.Am J Pathol. 2015 Aug;185(8):2105-17. doi: 10.1016/j.ajpath.2015.04.017. Epub 2015 Jun 13. Am J Pathol. 2015. PMID: 26079813 Free PMC article.
-
Suramin prevents the development of diabetic kidney disease by inhibiting NLRP3 inflammasome activation in KK-Ay mice.J Diabetes Investig. 2023 Feb;14(2):205-220. doi: 10.1111/jdi.13930. Epub 2022 Oct 29. J Diabetes Investig. 2023. PMID: 36308062 Free PMC article.
-
Astragaloside IV Alleviates Podocyte Injury in Diabetic Nephropathy through Regulating IRE-1α/NF-κ B/NLRP3 Pathway.Chin J Integr Med. 2024 Jul 23. doi: 10.1007/s11655-024-3568-0. Online ahead of print. Chin J Integr Med. 2024. PMID: 39039342
References
-
- Chow F, Ozols E, Nikolic-Paterson DJ, et al. Macrophages in mouse type 2 diabetic nephropathy: correlation with diabetic state and progressive renal injury. Kidney Int. 2004;65:116–128. - PubMed
-
- Nguyen D, Ping F, Mu W, et al. Macrophage accumulation in human progressive diabetic nephropathy. Nephrology (Carlton) 2006;11:226–231. - PubMed
-
- Lim AK, Ma FY, Nikolic-Paterson DJ, et al. Lymphocytes promote albuminuria, but not renal dysfunction or histological damage in a mouse model of diabetic renal injury. Diabetologia. 2010;53:1772–1782. - PubMed
-
- Moriya R, Manivel JC, Mauer M. Juxtaglomerular apparatus T-cell infiltration affects glomerular structure in Type 1 diabetic patients. Diabetologia. 2004;47:82–88. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
