

Structure of β-galactosidase at 3.2-Å resolution obtained by cryo-electron microscopy
- PMID: 25071206
- PMCID: PMC4136629
- DOI: 10.1073/pnas.1402809111
Structure of β-galactosidase at 3.2-Å resolution obtained by cryo-electron microscopy
Abstract
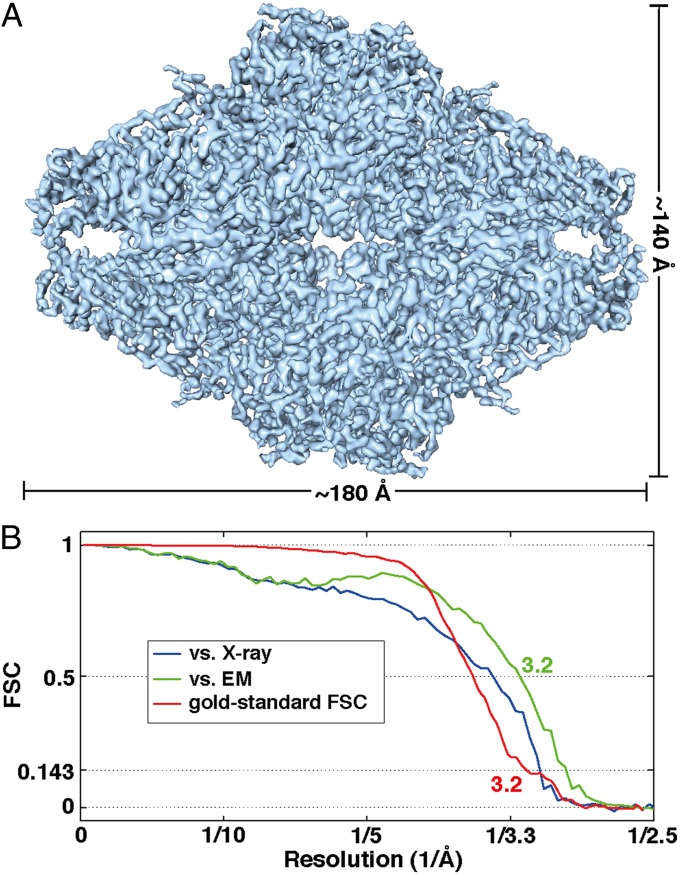
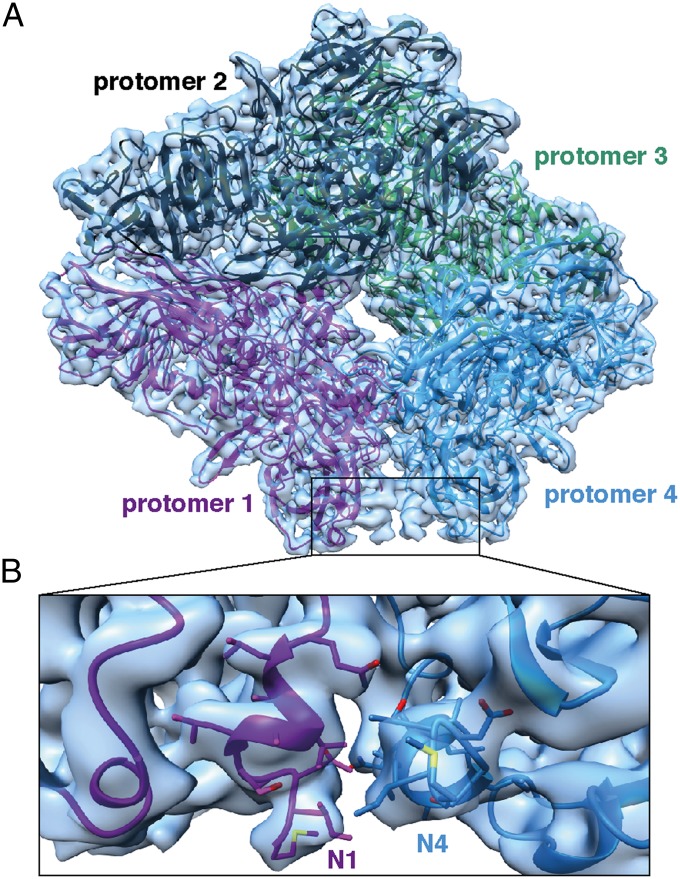
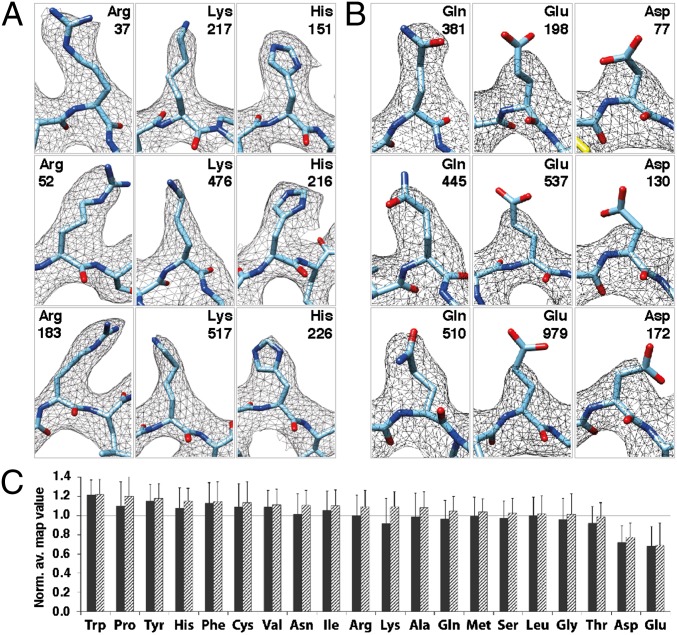
We report the solution structure of Escherichia coli β-galactosidase (∼465 kDa), solved at ∼3.2-Å resolution by using single-particle cryo-electron microscopy (cryo-EM). Densities for most side chains, including those of residues in the active site, and a catalytic Mg(2+) ion can be discerned in the map obtained by cryo-EM. The atomic model derived from our cryo-EM analysis closely matches the 1.7-Å crystal structure with a global rmsd of ∼0.66 Å. There are significant local differences throughout the protein, with clear evidence for conformational changes resulting from contact zones in the crystal lattice. Inspection of the map reveals that although densities for residues with positively charged and neutral side chains are well resolved, systematically weaker densities are observed for residues with negatively charged side chains. We show that the weaker densities for negatively charged residues arise from their greater sensitivity to radiation damage from electron irradiation as determined by comparison of density maps obtained by using electron doses ranging from 10 to 30 e(-)/Å(2). In summary, we establish that it is feasible to use cryo-EM to determine near-atomic resolution structures of protein complexes (<500 kDa) with low symmetry, and that the residue-specific radiation damage that occurs with increasing electron dose can be monitored by using dose fractionation tools available with direct electron detector technology.
Keywords: 3D reconstruction; CTF determination; frame alignment; single-particle EM; structure refinement.
Conflict of interest statement
The authors declare no conflict of interest.
Figures



Similar articles
-
2.2 Å resolution cryo-EM structure of β-galactosidase in complex with a cell-permeant inhibitor.Science. 2015 Jun 5;348(6239):1147-51. doi: 10.1126/science.aab1576. Epub 2015 May 7. Science. 2015. PMID: 25953817 Free PMC article.
-
Atomic Resolution Cryo-EM Structure of β-Galactosidase.Structure. 2018 Jun 5;26(6):848-856.e3. doi: 10.1016/j.str.2018.04.004. Epub 2018 May 10. Structure. 2018. PMID: 29754826 Free PMC article.
-
Determining the Crystal Structure of TRPV6.In: Kozak JA, Putney JW Jr, editors. Calcium Entry Channels in Non-Excitable Cells. Boca Raton (FL): CRC Press/Taylor & Francis; 2018. Chapter 14. In: Kozak JA, Putney JW Jr, editors. Calcium Entry Channels in Non-Excitable Cells. Boca Raton (FL): CRC Press/Taylor & Francis; 2018. Chapter 14. PMID: 30299652 Free Books & Documents. Review.
-
Beam-induced motion correction for sub-megadalton cryo-EM particles.Elife. 2014 Aug 13;3:e03665. doi: 10.7554/eLife.03665. Elife. 2014. PMID: 25122622 Free PMC article.
-
Processing of Structurally Heterogeneous Cryo-EM Data in RELION.Methods Enzymol. 2016;579:125-57. doi: 10.1016/bs.mie.2016.04.012. Epub 2016 May 31. Methods Enzymol. 2016. PMID: 27572726 Review.
Cited by
-
Comparative evaluation of the extracellular production of a polyethylene terephthalate degrading cutinase by Corynebacterium glutamicum and leaky Escherichia coli in batch and fed-batch processes.Microb Cell Fact. 2024 Oct 10;23(1):274. doi: 10.1186/s12934-024-02547-2. Microb Cell Fact. 2024. PMID: 39390488 Free PMC article.
-
Visualization of the Cdc48 AAA+ ATPase protein unfolding pathway.Nat Commun. 2024 Aug 29;15(1):7505. doi: 10.1038/s41467-024-51835-3. Nat Commun. 2024. PMID: 39209885 Free PMC article.
-
Nucleated synthetic cells with genetically driven intercompartment communication.Proc Natl Acad Sci U S A. 2024 Sep 3;121(36):e2404790121. doi: 10.1073/pnas.2404790121. Epub 2024 Aug 26. Proc Natl Acad Sci U S A. 2024. PMID: 39186653 Free PMC article.
-
Accurate size-based protein localization from cryo-ET tomograms.J Struct Biol X. 2024 Jun 26;10:100104. doi: 10.1016/j.yjsbx.2024.100104. eCollection 2024 Dec. J Struct Biol X. 2024. PMID: 39044770 Free PMC article.
-
Single molecule delivery into living cells.Nat Commun. 2024 May 23;15(1):4403. doi: 10.1038/s41467-024-48608-3. Nat Commun. 2024. PMID: 38782907 Free PMC article.
References
-
- Subramaniam S, Henderson R. Molecular mechanism of vectorial proton translocation by bacteriorhodopsin. Nature. 2000;406(6796):653–657. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
