

doi: 10.1016/j.cell.2014.06.037.
Conducting a microbiome study
Affiliations
- PMID: 25036628
- PMCID: PMC5074386
- DOI: 10.1016/j.cell.2014.06.037
Item in Clipboard
Conducting a microbiome study
Cell.
.
Abstract
Human microbiome research is an actively developing area of inquiry, with ramifications for our lifestyles, our interactions with microbes, and how we treat disease. Advances depend on carefully executed, controlled, and reproducible studies. Here, we provide a Primer for researchers from diverse disciplines interested in conducting microbiome research. We discuss factors to be considered in the design, execution, and data analysis of microbiome studies. These recommendations should help researchers to enter and contribute to this rapidly developing field.
Copyright © 2014 Elsevier Inc. All rights reserved.
Figures
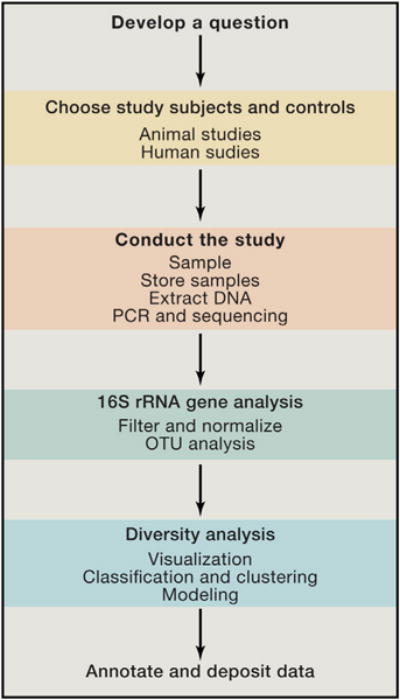
The sequential steps of conducting a microbiome study are diagramed, mirroring the sections of this Primer.
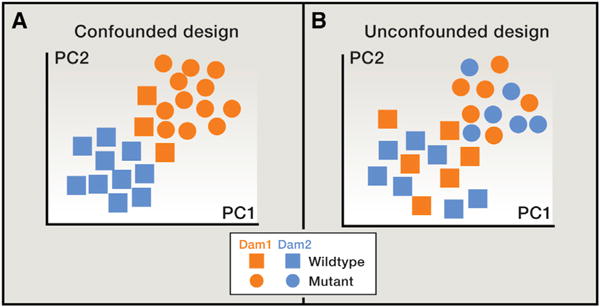
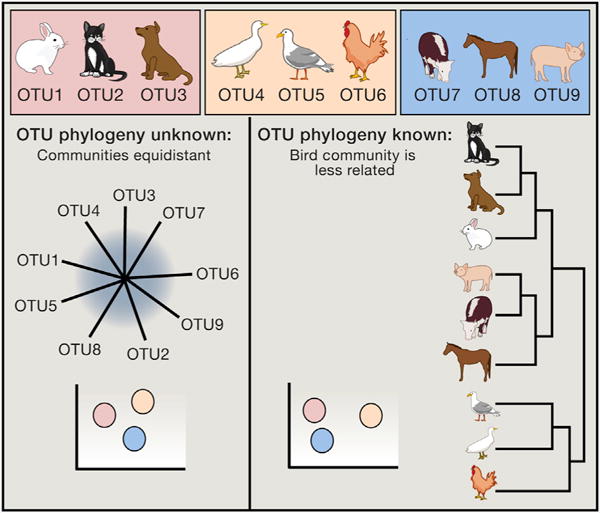
(A and B) In this mock example, each point represents a gut microbial community as characterized by a set of 16S rRNA gene sequences from a single mouse sample. In principal coordinates analysis (PCoA), points that are closer together represent microbial communities that are more similar in sequence composition. Samples from two different mouse genotypes are represented, and the mice are derived from two different dams. In all panels, squares indicate wild-type, and circles indicate mutant mouse genotypes. In (A), the effect of genotype is confounded by the effect of a shared dam, whereas in (B), the effect of dam is randomized across the two genotypes.
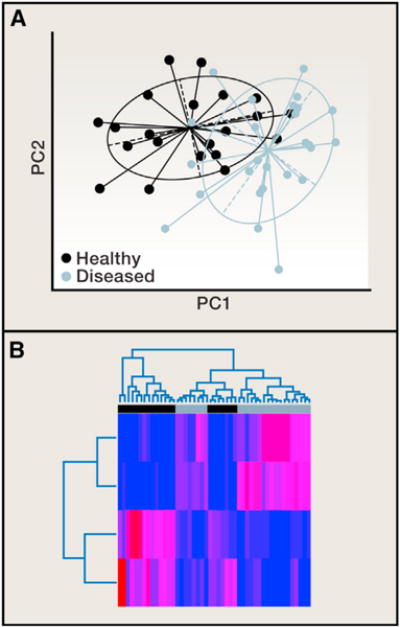
(A) Principal coordinates (PCs) from a principal coordinates analysis (PCoA) are plotted against each other to summarize the microbial community compositional differences between samples. Each point represents a single sample, and the distance between points represents how compositionally different the samples are from one another. The points are colored by health state, showing a clear difference in the microbial community composition between diseased (green) and healthy (purple). (B) Classification methods can be used to determine which OTUs discriminate between the healthy and diseased groups, and a heatmap can be used to visualize over/under representation of these OTUs in the groups. In this example, the abundances of the four discriminatory OTUs (rows) are colored from low abundance (blue) to high abundance (red) in the 47 samples (columns). Both the PCoA plot and the sample dendrogram in the heatmap show that the separation between disease and health states is not perfect. There is some overlap in the composition of these samples, though the placement of points in the PCoA plot is far from random. This observation should be supported with statistical analysis. For example, a Monte Carlo two-sample t test, comparing the distribution of within-group distances to the distribution of between-group distances applied to these data tells us that this clustering pattern is statistically significant.
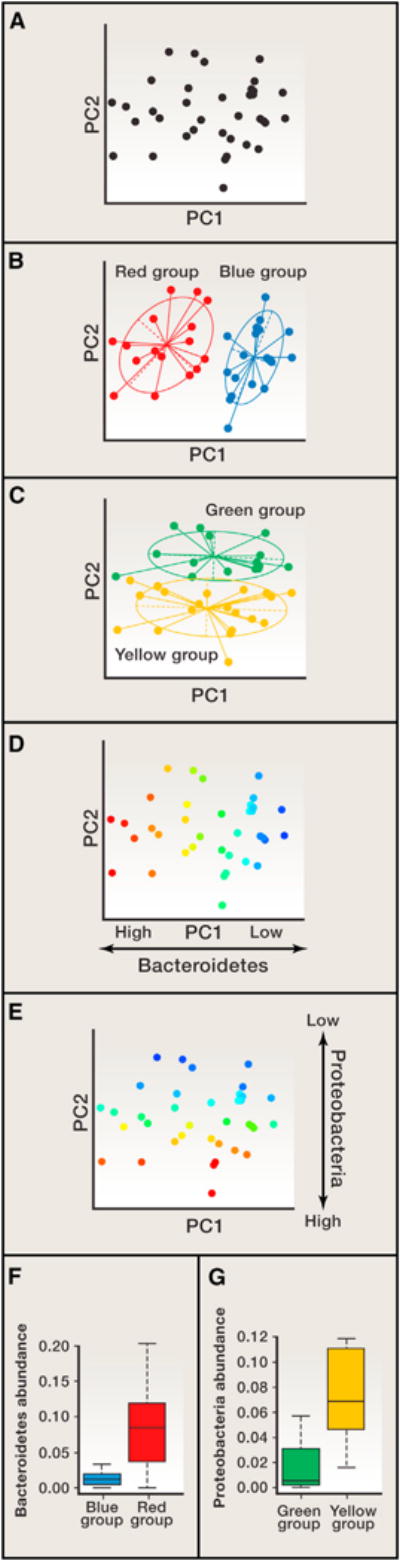
(A–C) In this simulated microbiome data set, a principal coordinates analysis (PCoA) was performed, and the first two principal coordinates, PC1 and PC2, are plotted. The exact same set of points is shown in panels (A–E) but is colored differently. In (A), samples are all colored black to show that they form gradients along PCs 1 and 2. In (B) and (C), two sets of clusters were designated by bisecting the spread of samples. In (B), half of the samples form the red cluster, and the second half form the Blue cluster along PC1. In (C), half of the samples are in the Green cluster, and the second half form the Yellow cluster along PC2. In (B) and (C), starplots display inferred clusters; this display can give the misleading impression of distinct clusters (see A; the data structure consists of gradients, not distinct clusters). (D–G) In (D) and (E), the samples are colored according to the abundances of the taxa that drive their separation along PCs 1 and 2. (D) The abundance of sequences belonging to the Bacteroidetes phylum drives the spread of samples along PC1; (E) abundances of Proteobacteria in the samples drive their spread along PC2. When the relative abundances for these phyla in samples are averaged (F and G), it is apparent that the Blue samples, which are at the “low end” of the Bacteroidetes gradient, have lower means than the Red samples, which are at the high end (F). Similarly, because the Yellow/Green samples are spread along PC2 according to their abundance of Proteobacteria, these two groups will also exhibit different mean abundances (G). Therefore, plotting mean values of the abundances of taxa that drive the gradients in the PCoA plots does not constitute a validation of the PCoA patterns.
Similar articles
-
Culture of previously uncultured members of the human gut microbiota by culturomics.Nat Microbiol. 2016 Nov 7;1:16203. doi: 10.1038/nmicrobiol.2016.203. Nat Microbiol. 2016. PMID: 27819657
-
Mining the Factors Driving the Evolution of the Pit Mud Microbiome under the Impact of Long-Term Production of Strong-Flavor Baijiu.Appl Environ Microbiol. 2021 Aug 11;87(17):e0088521. doi: 10.1128/AEM.00885-21. Epub 2021 Aug 11. Appl Environ Microbiol. 2021. PMID: 34160281 Free PMC article.
-
Profiling the microbial community of a Triassic halite deposit in Northern Ireland: an environment with significant potential for biodiscovery.FEMS Microbiol Lett. 2019 Nov 1;366(22):fnz242. doi: 10.1093/femsle/fnz242. FEMS Microbiol Lett. 2019. PMID: 31778179
-
Detection of micro-organisms in the environment.Biochem Soc Trans. 1995 May;23(2):435-7. doi: 10.1042/bst0230435. Biochem Soc Trans. 1995. PMID: 7672437 Review. No abstract available.
-
Molecular characterization of the human microbiome from a reproductive perspective.Fertil Steril. 2015 Dec;104(6):1344-50. doi: 10.1016/j.fertnstert.2015.10.008. Epub 2015 Oct 23. Fertil Steril. 2015. PMID: 26602982 Review.
Cited by
-
Changes in the Firmicutes to Bacteriodetes ratio in the gut microbiome in individuals with anorexia nervosa following inpatient treatment: A systematic review and a case series.Brain Behav. 2024 Sep;14(9):e70014. doi: 10.1002/brb3.70014. Brain Behav. 2024. PMID: 39295072 Free PMC article.
-
Clearing the plate: a strategic approach to mitigate well-to-well contamination in large-scale microbiome studies.mSystems. 2024 Oct 22;9(10):e0098524. doi: 10.1128/msystems.00985-24. Epub 2024 Sep 16. mSystems. 2024. PMID: 39283083 Free PMC article.
-
Bacteria, Fungi, and Scalp Psoriasis: Understanding the Role of the Microbiome in Disease Severity.J Clin Med. 2024 Aug 16;13(16):4846. doi: 10.3390/jcm13164846. J Clin Med. 2024. PMID: 39200988 Free PMC article.
-
Gut microbiota influences onset of foraging-related behavior but not physiological hallmarks of division of labor in honeybees.mBio. 2024 Sep 11;15(9):e0103424. doi: 10.1128/mbio.01034-24. Epub 2024 Jul 29. mBio. 2024. PMID: 39072646 Free PMC article.
-
Gut microbiome changes associated with chronic pancreatitis and pancreatic cancer: a systematic review and meta-analysis.Int J Surg. 2024 Sep 1;110(9):5781-5794. doi: 10.1097/JS9.0000000000001724. Int J Surg. 2024. PMID: 38847785 Free PMC article.
References
-
- Abarenkov K, Henrik Nilsson R, Larsson KH, Alexander IJ, Eberhardt U, Erland S, Høiland K, Kjøller R, Larsson E, Pennanen T, et al. The UNITE database for molecular identification of fungi—recent updates and future perspectives. New Phytol. 2010;186:281–285. - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
