

Linking the genetic architecture of cytosine modifications with human complex traits
- PMID: 24943591
- PMCID: PMC4204771
- DOI: 10.1093/hmg/ddu313
Linking the genetic architecture of cytosine modifications with human complex traits
Abstract
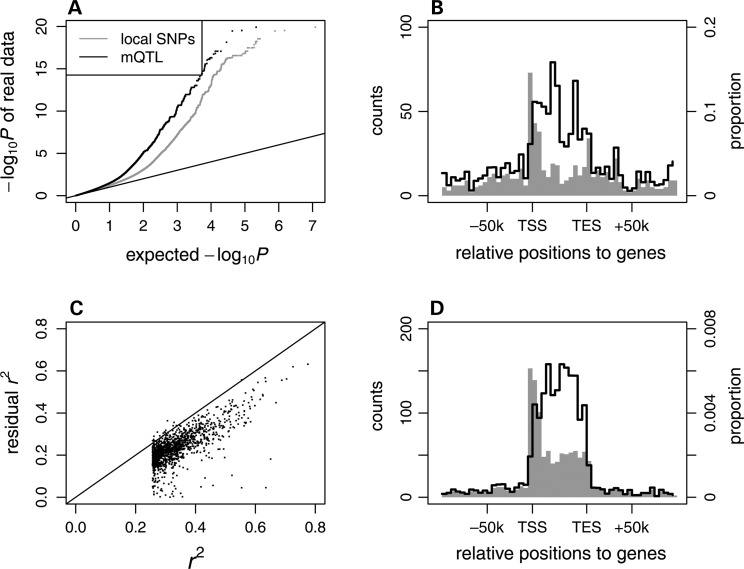
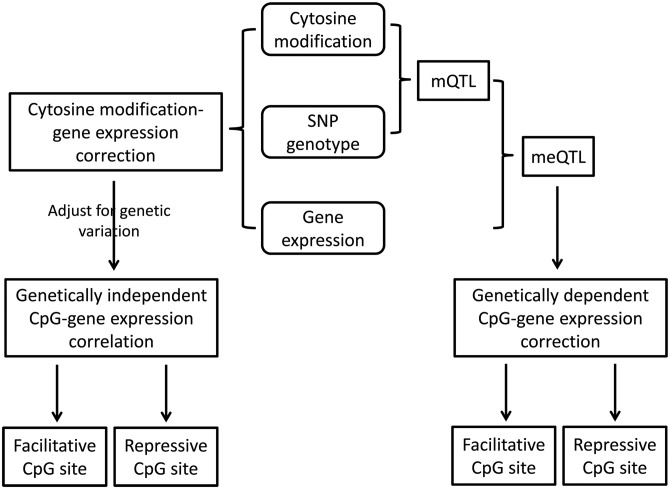
Interindividual variation in cytosine modifications could contribute to heterogeneity in disease risks and other complex traits. We assessed the genetic architecture of cytosine modifications at 283,540 CpG sites in lymphoblastoid cell lines (LCLs) derived from independent samples of European and African descent. Our study suggests that cytosine modification variation was primarily controlled in local by single major modification quantitative trait locus (mQTL) and additional minor loci. Local genetic epistasis was detectable for a small proportion of CpG sites, which were enriched by more than 9-fold for CpG sites mapped to population-specific mQTL. Genetically dependent CpG sites whose modification levels negatively (repressive sites) or positively (facilitative sites) correlated with gene expression levels significantly co-localized with transcription factor binding, with the repressive sites predominantly associated with active promoters whereas the facilitative sites rarely at active promoters. Genetically independent repressive or facilitative sites preferentially modulated gene expression variation by influencing local chromatin accessibility, with the facilitative sites primarily antagonizing H3K27me3 and H3K9me3 deposition. In comparison with expression quantitative trait loci (eQTL), mQTL detected from LCLs were enriched in associations for a broader range of disease categories including chronic inflammatory, autoimmune and psychiatric disorders, suggesting that cytosine modification variation, while possesses a degree of cell linage specificity, is more stably inherited over development than gene expression variation. About 11% of unique single-nucleotide polymorphisms reported in the Genome-Wide Association Study Catalog were annotated, 78% as mQTL and 31% as eQTL in LCLs, which covered 37% of the investigated diseases/traits and provided insights to the biological mechanisms.
© The Author 2014. Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures



Similar articles
-
Linking short tandem repeat polymorphisms with cytosine modifications in human lymphoblastoid cell lines.Hum Genet. 2016 Feb;135(2):223-32. doi: 10.1007/s00439-015-1628-4. Epub 2015 Dec 30. Hum Genet. 2016. PMID: 26714498 Free PMC article.
-
Genome-wide variation of cytosine modifications between European and African populations and the implications for complex traits.Genetics. 2013 Aug;194(4):987-96. doi: 10.1534/genetics.113.151381. Epub 2013 Jun 21. Genetics. 2013. PMID: 23792949 Free PMC article.
-
Transcript Isoform Variation Associated with Cytosine Modification in Human Lymphoblastoid Cell Lines.Genetics. 2016 Jun;203(2):985-95. doi: 10.1534/genetics.115.185504. Epub 2016 Mar 30. Genetics. 2016. PMID: 27029734 Free PMC article.
-
Studying the epigenome using next generation sequencing.J Med Genet. 2011 Nov;48(11):721-30. doi: 10.1136/jmedgenet-2011-100242. Epub 2011 Aug 8. J Med Genet. 2011. PMID: 21825079 Review.
-
Expression Quantitative Trait Loci Information Improves Predictive Modeling of Disease Relevance of Non-Coding Genetic Variation.PLoS One. 2015 Oct 16;10(10):e0140758. doi: 10.1371/journal.pone.0140758. eCollection 2015. PLoS One. 2015. PMID: 26474488 Free PMC article. Review.
Cited by
-
Associations of MTHFR Polymorphisms and Cytosine Modifications with Early-Gestational Diabetes Mellitus in Chinese Pregnant Women.Reprod Sci. 2023 Oct;30(10):2973-2982. doi: 10.1007/s43032-023-01247-3. Epub 2023 May 8. Reprod Sci. 2023. PMID: 37154866
-
Differential methylation in EGLN1 associates with blood oxygen saturation and plasma protein levels in high-altitude pulmonary edema.Clin Epigenetics. 2022 Sep 30;14(1):123. doi: 10.1186/s13148-022-01338-z. Clin Epigenetics. 2022. PMID: 36180894 Free PMC article.
-
Deep learning predicts DNA methylation regulatory variants in the human brain and elucidates the genetics of psychiatric disorders.Proc Natl Acad Sci U S A. 2022 Aug 23;119(34):e2206069119. doi: 10.1073/pnas.2206069119. Epub 2022 Aug 15. Proc Natl Acad Sci U S A. 2022. PMID: 35969790 Free PMC article.
-
Variants in FtsJ RNA 2'-O-Methyltransferase 3 and Growth Hormone 1 are associated with small body size and a dental anomaly in dogs.Proc Natl Acad Sci U S A. 2020 Oct 6;117(40):24929-24935. doi: 10.1073/pnas.2009500117. Epub 2020 Sep 21. Proc Natl Acad Sci U S A. 2020. PMID: 32958658 Free PMC article.
-
DNA hypermethylation in disease: mechanisms and clinical relevance.Epigenetics. 2019 Dec;14(12):1141-1163. doi: 10.1080/15592294.2019.1638701. Epub 2019 Jul 8. Epigenetics. 2019. PMID: 31284823 Free PMC article. Review.
References
-
- HapMap. The international HapMap project. Nature. 2003;426:789–796. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
