

Ketogenic diets, mitochondria, and neurological diseases
- PMID: 24847102
- PMCID: PMC4617125
- DOI: 10.1194/jlr.R048975
Ketogenic diets, mitochondria, and neurological diseases
Abstract
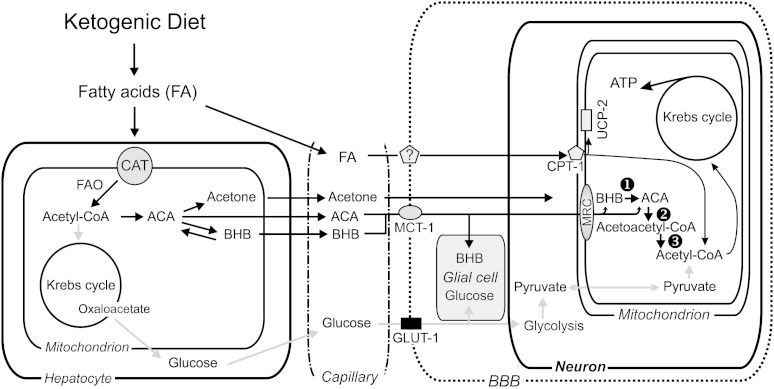
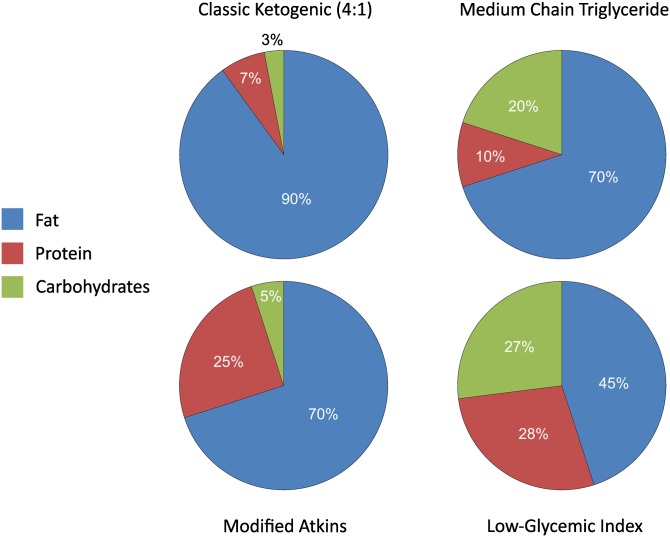
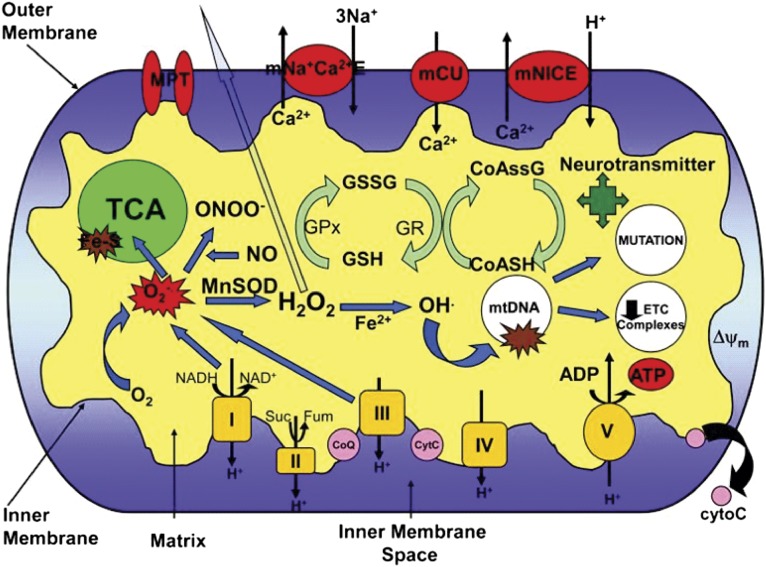
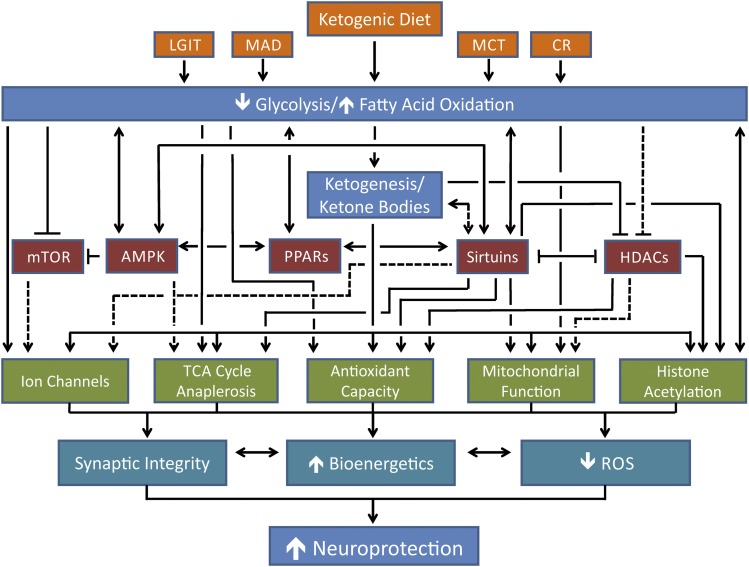
The ketogenic diet (KD) is a broad-spectrum therapy for medically intractable epilepsy and is receiving growing attention as a potential treatment for neurological disorders arising in part from bioenergetic dysregulation. The high-fat/low-carbohydrate "classic KD", as well as dietary variations such as the medium-chain triglyceride diet, the modified Atkins diet, the low-glycemic index treatment, and caloric restriction, enhance cellular metabolic and mitochondrial function. Hence, the broad neuroprotective properties of such therapies may stem from improved cellular metabolism. Data from clinical and preclinical studies indicate that these diets restrict glycolysis and increase fatty acid oxidation, actions which result in ketosis, replenishment of the TCA cycle (i.e., anaplerosis), restoration of neurotransmitter and ion channel function, and enhanced mitochondrial respiration. Further, there is mounting evidence that the KD and its variants can impact key signaling pathways that evolved to sense the energetic state of the cell, and that help maintain cellular homeostasis. These pathways, which include PPARs, AMP-activated kinase, mammalian target of rapamycin, and the sirtuins, have all been recently implicated in the neuroprotective effects of the KD. Further research in this area may lead to future therapeutic strategies aimed at mimicking the pleiotropic neuroprotective effects of the KD.
Keywords: cellular signaling; fatty acids; ketone; oxidative stress.
Copyright © 2014 by the American Society for Biochemistry and Molecular Biology, Inc.
Figures
Similar articles
-
Neuroketotherapeutics: A modern review of a century-old therapy.Neurochem Int. 2018 Jul;117:114-125. doi: 10.1016/j.neuint.2017.05.019. Epub 2017 Jun 1. Neurochem Int. 2018. PMID: 28579059 Free PMC article. Review.
-
[Ketogenic diet – mechanism of action and perspectives for the use in the therapy: data from clinical studies].Postepy Biochem. 2020 Dec 13;66(3):270-286. doi: 10.18388/pb.2020_342. Print 2020 Sep 30. Postepy Biochem. 2020. PMID: 33315315 Review. Polish.
-
Does the ketogenic ratio matter when using ketogenic diet therapy in pediatric epilepsy?Epilepsia. 2023 Feb;64(2):284-291. doi: 10.1111/epi.17476. Epub 2022 Dec 15. Epilepsia. 2023. PMID: 36471628 Review.
-
Therapeutic potential of the ketogenic diet: A metabolic switch with implications for neurological disorders, the gut-brain axis, and cardiovascular diseases.J Nutr Biochem. 2024 Oct;132:109693. doi: 10.1016/j.jnutbio.2024.109693. Epub 2024 Jun 14. J Nutr Biochem. 2024. PMID: 38880191 Review.
-
Activation of G protein-coupled receptors by ketone bodies: Clinical implication of the ketogenic diet in metabolic disorders.Front Endocrinol (Lausanne). 2022 Oct 20;13:972890. doi: 10.3389/fendo.2022.972890. eCollection 2022. Front Endocrinol (Lausanne). 2022. PMID: 36339405 Free PMC article. Review.
Cited by
-
Mechanism of Action of Ketogenic Diet Treatment: Impact of Decanoic Acid and Beta-Hydroxybutyrate on Sirtuins and Energy Metabolism in Hippocampal Murine Neurons.Nutrients. 2020 Aug 8;12(8):2379. doi: 10.3390/nu12082379. Nutrients. 2020. PMID: 32784510 Free PMC article.
-
Ketogenic Diet Has Moderate Effects on the Fecal Microbiota of Wild-Type Mice.Nutrients. 2023 Oct 31;15(21):4629. doi: 10.3390/nu15214629. Nutrients. 2023. PMID: 37960282 Free PMC article.
-
Ketogenic diet modifies the gut microbiota in a murine model of autism spectrum disorder.Mol Autism. 2016 Sep 1;7(1):37. doi: 10.1186/s13229-016-0099-3. eCollection 2016. Mol Autism. 2016. PMID: 27594980 Free PMC article.
-
Ketogenic Diet in Alzheimer's Disease.Int J Mol Sci. 2019 Aug 9;20(16):3892. doi: 10.3390/ijms20163892. Int J Mol Sci. 2019. PMID: 31405021 Free PMC article. Review.
-
Glycolytic inhibition: A novel approach toward controlling neuronal excitability and seizures.Epilepsia Open. 2018 Aug 19;3(Suppl Suppl 2):191-197. doi: 10.1002/epi4.12251. eCollection 2018 Dec. Epilepsia Open. 2018. PMID: 30564778 Free PMC article.
References
-
- Vining E. P., Freeman J. M., Ballaban-Gil K., Camfield C. S., Camfield P. R., Holmes G. L., Shinnar S., Shuman R., Trevathan E., Wheless J. W. 1998. A multicenter study of the efficacy of the ketogenic diet. Arch. Neurol. 55: 1433–1437. - PubMed
-
- Neal E. G., Chaffe H., Schwartz R. H., Lawson M. S., Edwards N., Fitzsimmons G., Whitney A., Cross J. H. 2008. The ketogenic diet for the treatment of childhood epilepsy: a randomised controlled trial. Lancet Neurol. 7: 500–506. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
