

Structural basis for protein antiaggregation activity of the trigger factor chaperone
- PMID: 24812405
- PMCID: PMC4070327
- DOI: 10.1126/science.1250494
Structural basis for protein antiaggregation activity of the trigger factor chaperone
Abstract
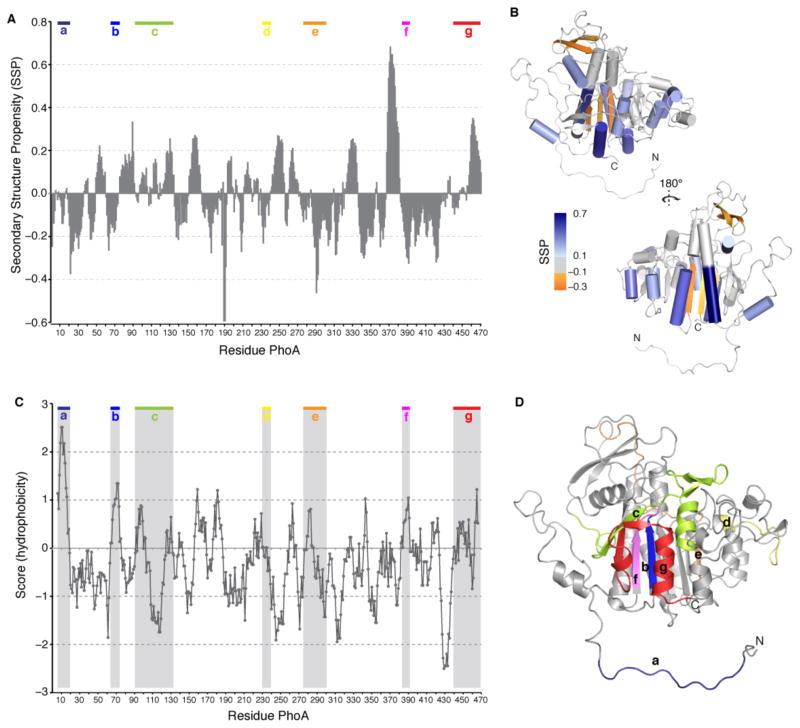
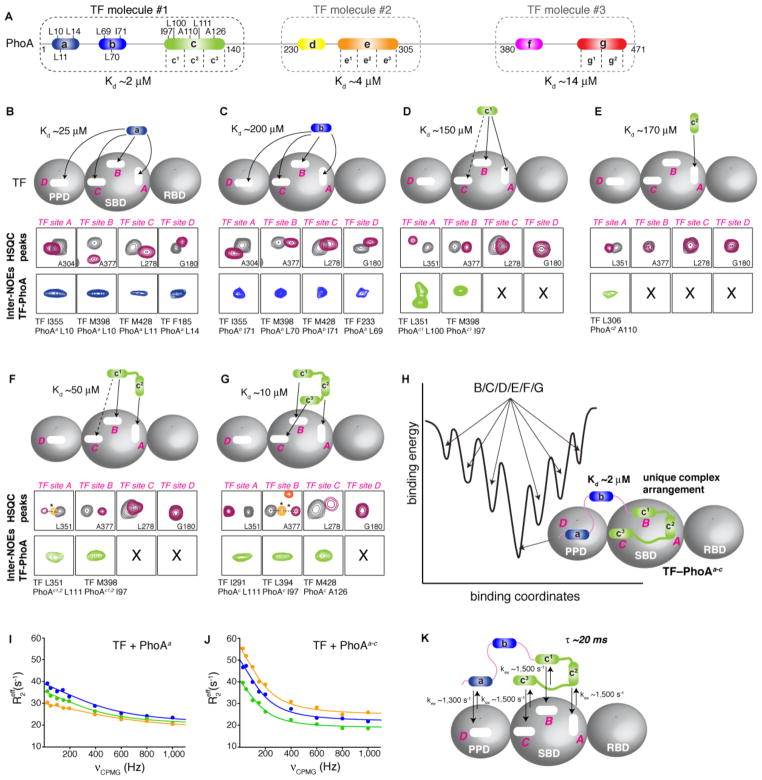
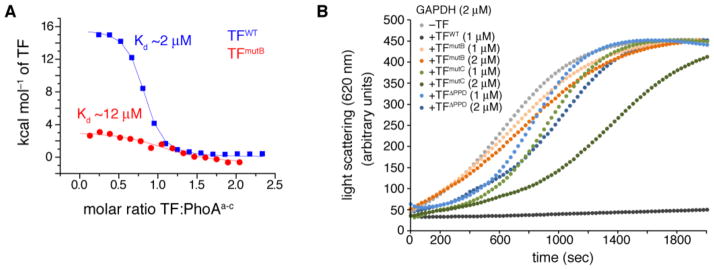
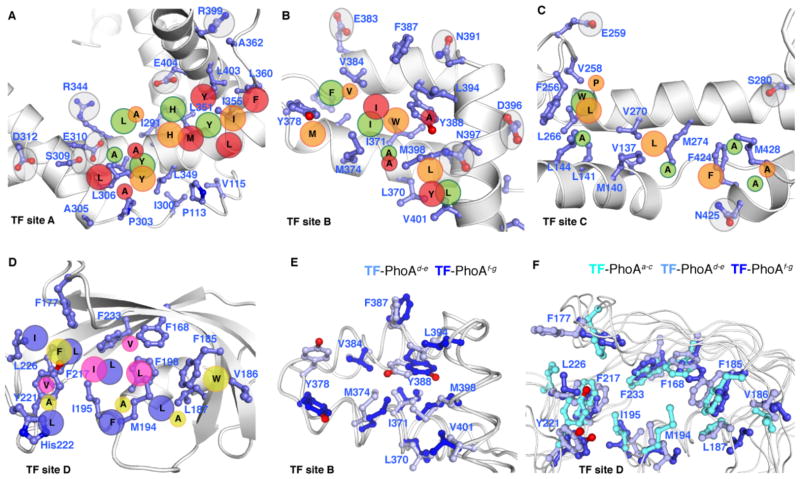
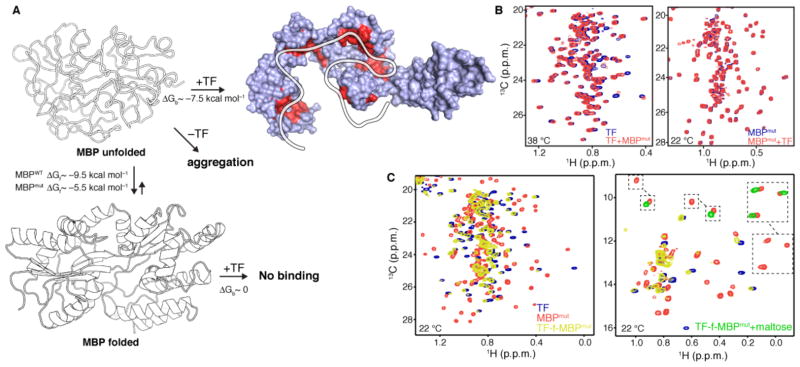
Molecular chaperones prevent aggregation and misfolding of proteins, but scarcity of structural data has impeded an understanding of the recognition and antiaggregation mechanisms. We report the solution structure, dynamics, and energetics of three trigger factor (TF) chaperone molecules in complex with alkaline phosphatase (PhoA) captured in the unfolded state. Our data show that TF uses multiple sites to bind to several regions of the PhoA substrate protein primarily through hydrophobic contacts. Nuclear magnetic resonance (NMR) relaxation experiments show that TF interacts with PhoA in a highly dynamic fashion, but as the number and length of the PhoA regions engaged by TF increase, a more stable complex gradually emerges. Multivalent binding keeps the substrate protein in an extended, unfolded conformation. The results show how molecular chaperones recognize unfolded polypeptides and, by acting as unfoldases and holdases, prevent the aggregation and premature (mis)folding of unfolded proteins.
Figures


Comment in
-
Biochemistry. Trigger factor flexibility.Science. 2014 May 9;344(6184):590-1. doi: 10.1126/science.1254064. Science. 2014. PMID: 24812390 No abstract available.
Similar articles
-
Biochemistry. Trigger factor flexibility.Science. 2014 May 9;344(6184):590-1. doi: 10.1126/science.1254064. Science. 2014. PMID: 24812390 No abstract available.
-
Structural basis for the antifolding activity of a molecular chaperone.Nature. 2016 Sep 8;537(7619):202-206. doi: 10.1038/nature18965. Epub 2016 Aug 8. Nature. 2016. PMID: 27501151 Free PMC article.
-
Structural insight into proline cis/trans isomerization of unfolded proteins catalyzed by the trigger factor chaperone.J Biol Chem. 2018 Sep 28;293(39):15095-15106. doi: 10.1074/jbc.RA118.003579. Epub 2018 Aug 9. J Biol Chem. 2018. PMID: 30093407 Free PMC article.
-
A cradle for new proteins: trigger factor at the ribosome.Curr Opin Struct Biol. 2005 Apr;15(2):204-12. doi: 10.1016/j.sbi.2005.03.005. Curr Opin Struct Biol. 2005. PMID: 15837180 Review.
-
Substrate Interaction Networks of the Escherichia coli Chaperones: Trigger Factor, DnaK and GroEL.Adv Exp Med Biol. 2015;883:271-94. doi: 10.1007/978-3-319-23603-2_15. Adv Exp Med Biol. 2015. PMID: 26621473 Review.
Cited by
-
Aha1 Exhibits Distinctive Dynamics Behavior and Chaperone-Like Activity.Molecules. 2021 Mar 30;26(7):1943. doi: 10.3390/molecules26071943. Molecules. 2021. PMID: 33808352 Free PMC article.
-
Simultaneous sensing and imaging of individual biomolecular complexes enabled by modular DNA-protein coupling.Commun Chem. 2020 Feb 12;3(1):20. doi: 10.1038/s42004-020-0267-4. Commun Chem. 2020. PMID: 36703465 Free PMC article.
-
Assessing Site-Specific Enhancements Imparted by Hyperpolarized Water in Folded and Unfolded Proteins by 2D HMQC NMR.J Am Chem Soc. 2020 May 20;142(20):9267-9284. doi: 10.1021/jacs.0c00807. Epub 2020 May 12. J Am Chem Soc. 2020. PMID: 32338002 Free PMC article.
-
The chaperone toolbox at the single-molecule level: From clamping to confining.Protein Sci. 2017 Jul;26(7):1291-1302. doi: 10.1002/pro.3161. Epub 2017 Apr 20. Protein Sci. 2017. PMID: 28342267 Free PMC article. Review.
-
PDI Family Members as Guides for Client Folding and Assembly.Int J Mol Sci. 2020 Dec 8;21(24):9351. doi: 10.3390/ijms21249351. Int J Mol Sci. 2020. PMID: 33302492 Free PMC article. Review.
References
-
- Bukau B, Weissman J, Horwich A. Molecular chaperones and protein quality control. Cell. 2006;125:443–451. - PubMed
-
- Kramer G, Boehringer D, Ban N, Bukau B. The ribosome as a platform for co-translational processing, folding and targeting of newly synthesized proteins. Nat Struct Mol Biol. 2009;16:589–597. - PubMed
-
- Ulrich Hartl F, Hayer-Hartl M. Converging concepts of protein folding in vitro and in vivo. Nat Struct Mol Biol. 2009;16:574–581. - PubMed
-
- Horwich AL. Protein folding in the cell: an inside story. Nat Med. 2011;17:1211–1216. - PubMed
-
- Hartl FU, Bracher A, Hayer-Hartl M. Molecular chaperones in protein folding and proteostasis. Nature. 2011;475:324–332. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
