

Phenotype and frequency of STUB1 mutations: next-generation screenings in Caucasian ataxia and spastic paraplegia cohorts
- PMID: 24742043
- PMCID: PMC4021831
- DOI: 10.1186/1750-1172-9-57
Phenotype and frequency of STUB1 mutations: next-generation screenings in Caucasian ataxia and spastic paraplegia cohorts
Abstract
Background: Mutations in the gene STUB1, encoding the protein CHIP (C-terminus of HSC70-interacting protein), have recently been suggested as a cause of recessive ataxia based on the findings in few Chinese families. Here we aimed to investigate the phenotypic and genotypic spectrum of STUB1 mutations, and to assess their frequency in different Caucasian disease cohorts.
Methods: 300 subjects with degenerative ataxia (n = 167) or spastic paraplegia (n = 133) were screened for STUB1 variants by whole-exome-sequencing (n = 204) or shotgun-fragment-library-sequencing (n = 96). To control for the specificity of STUB1 variants, we screened an additional 1707 exomes from 891 index families with other neurological diseases.
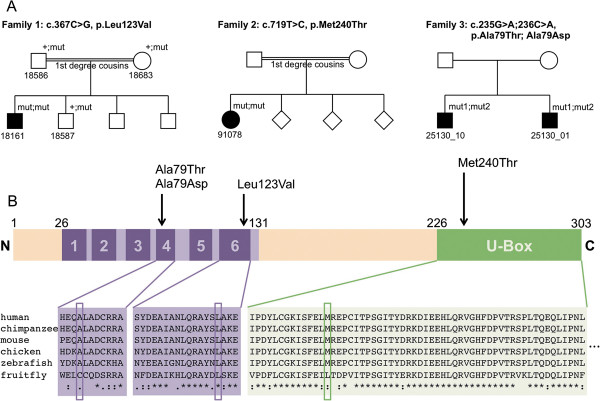
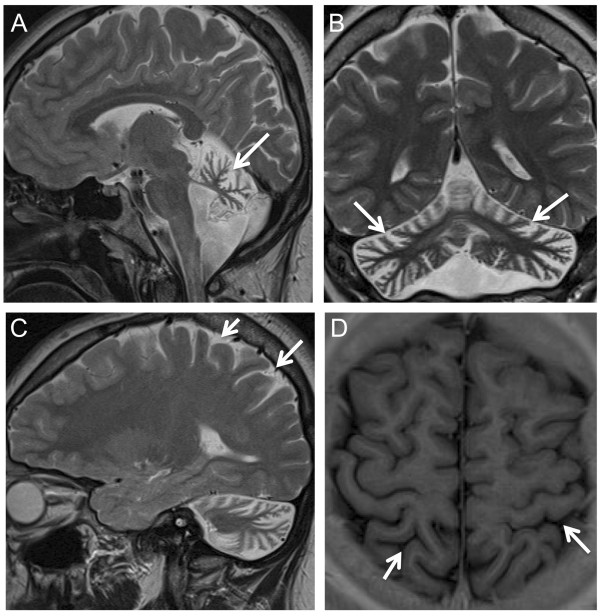
Results: We identified 3 ataxia patients (3/167 = 1.8%) with 4 novel missense mutations in STUB1, including 3 mutations in its tetratricopeptide-repeat domain. All patients showed evidence of pyramidal tract damage. Cognitive impairment was present only in one and hypogonadism in none of them. Ataxia did not start before age 48 years in one subject. No recessive STUB1 variants were identified in families with other neurological diseases, demonstrating that STUB1 variants are not simply rare polymorphisms ubiquitous in neurodegenerative disease.
Conclusions: STUB1-disease occurs also in Caucasian ataxia populations (1.8%). Our results expand the genotypic spectrum of STUB1-disease, showing that pathogenic mutations affect also the tetratricopeptide-repeat domain, thus providing clinical evidence for the functional importance of this domain. Moreover, they further delineate the phenotypic core features of STUB1-ataxia. Pyramidal tract damage is a common accompanying feature and can include lower limb spasticity, thus adding STUB1-ataxia to the differential diagnosis of "spastic ataxias". However, STUB1 is rare in subjects with predominant spastic paraplegia (0/133). In contrast to previous reports, STUB1-ataxia can start even above age 40 years, and neither hypogonadism nor prominent cognitive impairment are obligatory features.
Figures
Similar articles
-
STUB1/CHIP mutations cause Gordon Holmes syndrome as part of a widespread multisystemic neurodegeneration: evidence from four novel mutations.Orphanet J Rare Dis. 2017 Feb 13;12(1):31. doi: 10.1186/s13023-017-0580-x. Orphanet J Rare Dis. 2017. PMID: 28193273 Free PMC article.
-
STUB1 mutations in autosomal recessive ataxias - evidence for mutation-specific clinical heterogeneity.Orphanet J Rare Dis. 2014 Sep 26;9:146. doi: 10.1186/s13023-014-0146-0. Orphanet J Rare Dis. 2014. PMID: 25258038 Free PMC article.
-
A de novo STUB1 variant associated with an early adult-onset multisystemic ataxia phenotype.J Neurol. 2021 Oct;268(10):3845-3851. doi: 10.1007/s00415-021-10524-7. Epub 2021 Apr 3. J Neurol. 2021. PMID: 33811518 Free PMC article.
-
Spinocerebellar ataxia type 48: last but not least.Neurol Sci. 2020 Sep;41(9):2423-2432. doi: 10.1007/s10072-020-04408-3. Epub 2020 Apr 27. Neurol Sci. 2020. PMID: 32342324 Review.
-
Spastic paraplegia type 46: novel and recurrent GBA2 gene variants in a compound heterozygous Italian patient with spastic ataxia phenotype.Neurol Sci. 2021 Nov;42(11):4741-4745. doi: 10.1007/s10072-021-05463-0. Epub 2021 Jul 12. Neurol Sci. 2021. PMID: 34251556 Review.
Cited by
-
FAHN/SPG35: a narrow phenotypic spectrum across disease classifications.Brain. 2019 Jun 1;142(6):1561-1572. doi: 10.1093/brain/awz102. Brain. 2019. PMID: 31135052 Free PMC article.
-
SKELETAL MUSCLE MITOCHONDRIAL ALTERATIONS IN CARBOXYL TERMINUS OF HSC70 INTERACTING PROTEIN (CHIP) -/- MICE.Afr J Cell Pathol. 2016 Apr;6(4):28-36. Afr J Cell Pathol. 2016. PMID: 28593200 Free PMC article.
-
The molecular basis of spinocerebellar ataxia type 48 caused by a de novo mutation in the ubiquitin ligase CHIP.J Biol Chem. 2022 May;298(5):101899. doi: 10.1016/j.jbc.2022.101899. Epub 2022 Apr 7. J Biol Chem. 2022. PMID: 35398354 Free PMC article.
-
Extending the Phenotypic Spectrum Associated with STUB1 Mutations: A Case of Dystonia.Mov Disord Clin Pract. 2020 Mar 9;7(3):318-324. doi: 10.1002/mdc3.12914. eCollection 2020 Apr. Mov Disord Clin Pract. 2020. PMID: 32258232 Free PMC article.
-
CHIP mutations affect the heat shock response differently in human fibroblasts and iPSC-derived neurons.Dis Model Mech. 2020 Oct 12;13(10):dmm045096. doi: 10.1242/dmm.045096. Dis Model Mech. 2020. PMID: 33097556 Free PMC article.
References
-
- Synofzik M, Schöls L, Rieß O. Hereditäre Ataxien. Aktuelle Übersicht und diagnostische Strategien Medizinische Genetik; 2013. pp. 235–248.
-
- Shi CH, Schisler JC, Rubel CE, Tan S, Song B, McDonough H, Xu L, Portbury AL, Mao CY, True C, Wang RH, Wang QZ, Sun SL, Seminara SB, Patterson C, Xu YM. Ataxia and hypogonadism caused by the loss of ubiquitin ligase activity of the U box protein CHIP. Hum Mol Genet. 2014;23:1013–1024. doi: 10.1093/hmg/ddt497. - DOI - PMC - PubMed
-
- Shi Y, Wang J, Li JD, Ren H, Guan W, He M, Yan W, Zhou Y, Hu Z, Zhang J, Xiao J, Su Z, Dai M, Wang J, Jiang H, Guo J, Zhou Y, Zhang F, Li N, Du J, Xu Q, Hu Y, Pan Q, Shen L, Wang G, Xia K, Zhang Z, Tang B. Identification of CHIP as a novel causative gene for autosomal recessive cerebellar ataxia. PLoS One. 2013;8:e81884. doi: 10.1371/journal.pone.0081884. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
