

Indoleamine 2,3-dioxygenase pathways of pathogenic inflammation and immune escape in cancer
- PMID: 24711084
- PMCID: PMC4384696
- DOI: 10.1007/s00262-014-1549-4
Indoleamine 2,3-dioxygenase pathways of pathogenic inflammation and immune escape in cancer
Abstract
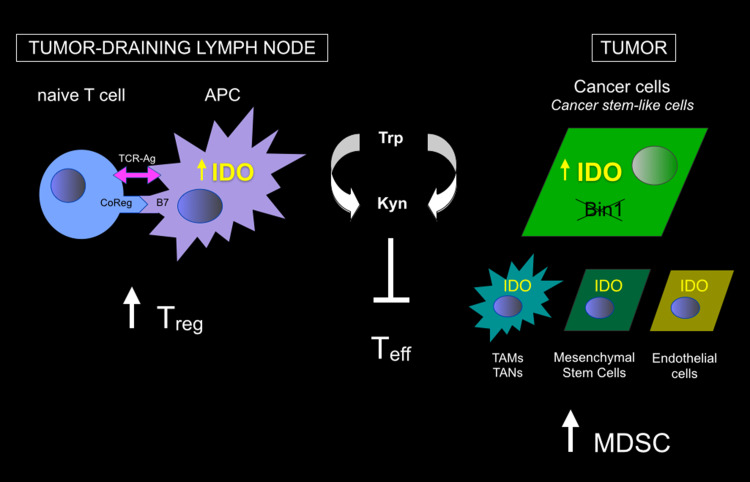
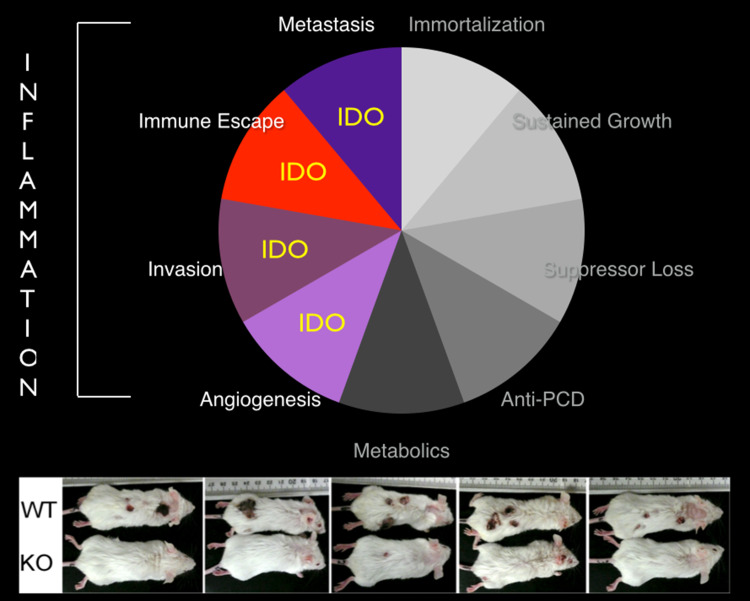
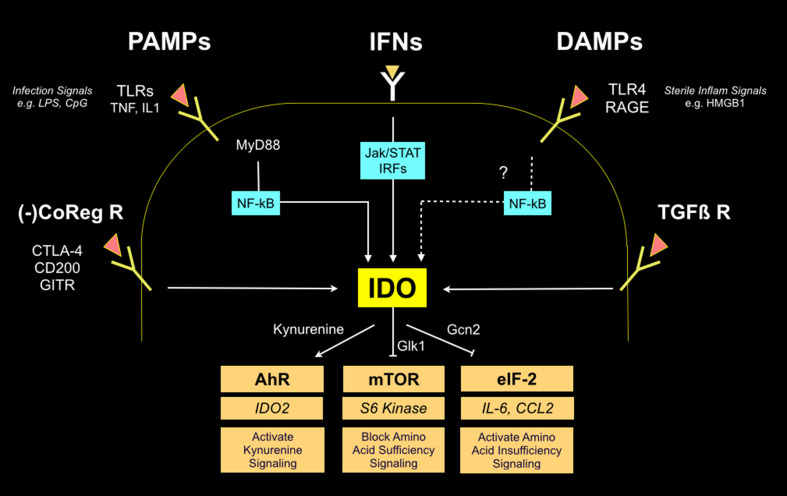
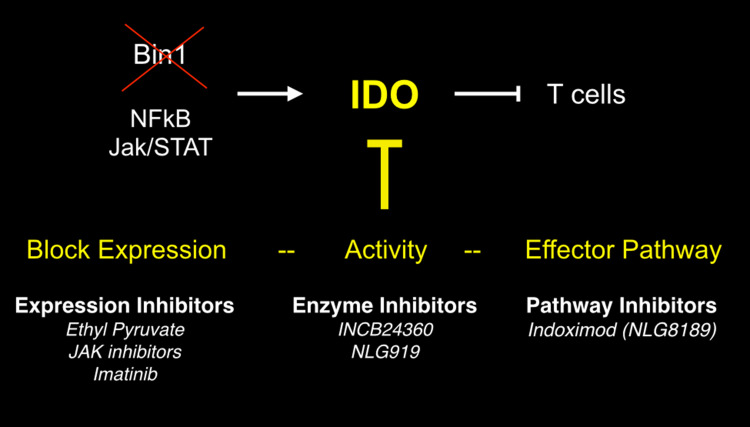
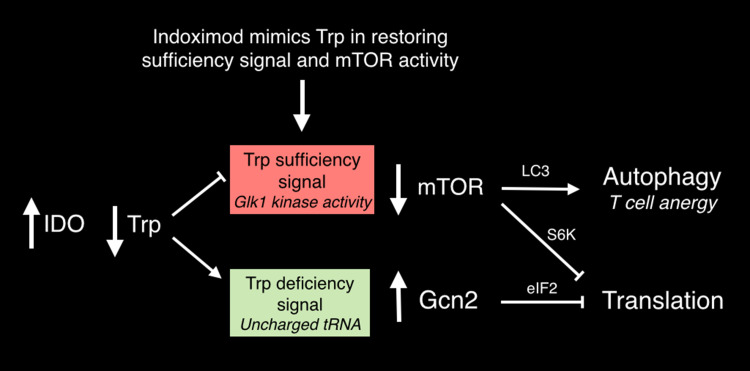
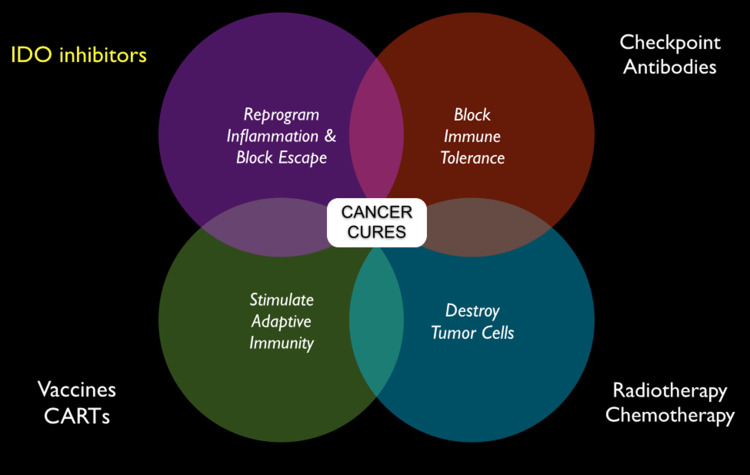
Genetic and pharmacological studies of indoleamine 2,3-dioxygenase (IDO) have established this tryptophan catabolic enzyme as a central driver of malignant development and progression. IDO acts in tumor, stromal and immune cells to support pathogenic inflammatory processes that engender immune tolerance to tumor antigens. The multifaceted effects of IDO activation in cancer include the suppression of T and NK cells, the generation and activation of T regulatory cells and myeloid-derived suppressor cells, and the promotion of tumor angiogenesis. Mechanistic investigations have defined the aryl hydrocarbon receptor, the master metabolic regulator mTORC1 and the stress kinase Gcn2 as key effector signaling elements for IDO, which also exerts a non-catalytic role in TGF-β signaling. Small-molecule inhibitors of IDO exhibit anticancer activity and cooperate with immunotherapy, radiotherapy or chemotherapy to trigger rapid regression of aggressive tumors otherwise resistant to treatment. Notably, the dramatic antitumor activity of certain targeted therapeutics such as imatinib (Gleevec) in gastrointestinal stromal tumors has been traced in part to IDO downregulation. Further, antitumor responses to immune checkpoint inhibitors can be heightened safely by a clinical lead inhibitor of the IDO pathway that relieves IDO-mediated suppression of mTORC1 in T cells. In this personal perspective on IDO as a nodal mediator of pathogenic inflammation and immune escape in cancer, we provide a conceptual foundation for the clinical development of IDO inhibitors as a novel class of immunomodulators with broad application in the treatment of advanced human cancer.
Conflict of interest statement
G.C. Prendergast, R. Metz and A.J. Muller state a conflict of interest as shareholders and G.C. Prendergast also a grant recipient and a member of the scientific advisory board for New Link Genetics Corporation, a biopharmaceutical company that has licensed IDO intellectual property for clinical development from the Lankenau Institute of Medical Research, as described in U.S. Patents Nos. 7705022, 7714139, 8008281, 8058416, 8383613, 8389568, 8436151, 8476454 and 8586636. The other authors state no conflict of interest.
Figures
Similar articles
-
Indoleamine 2,3-dioxygenase in T-cell tolerance and tumoral immune escape.Immunol Rev. 2008 Apr;222:206-21. doi: 10.1111/j.1600-065X.2008.00610.x. Immunol Rev. 2008. PMID: 18364004 Review.
-
Indoleamine 2,3-dioxygenase: is it an immune suppressor?Cancer J. 2010 Jul-Aug;16(4):354-9. doi: 10.1097/PPO.0b013e3181eb3343. Cancer J. 2010. PMID: 20693847 Free PMC article. Review.
-
Indoleamine 2,3-dioxygenase and dendritic cell tolerogenicity.Immunol Invest. 2012;41(6-7):738-64. doi: 10.3109/08820139.2012.676122. Immunol Invest. 2012. PMID: 23017144 Free PMC article. Review.
-
Targeting indoleamine-2,3-dioxygenase in cancer: Scientific rationale and clinical evidence.Pharmacol Ther. 2019 Apr;196:105-116. doi: 10.1016/j.pharmthera.2018.12.004. Epub 2018 Dec 4. Pharmacol Ther. 2019. PMID: 30521884 Review.
-
Indoleamine 2,3-Dioxygenase and Its Therapeutic Inhibition in Cancer.Int Rev Cell Mol Biol. 2018;336:175-203. doi: 10.1016/bs.ircmb.2017.07.004. Epub 2017 Sep 21. Int Rev Cell Mol Biol. 2018. PMID: 29413890 Free PMC article. Review.
Cited by
-
Differential Roles of IDO1 and IDO2 in T and B Cell Inflammatory Immune Responses.Front Immunol. 2020 Aug 18;11:1861. doi: 10.3389/fimmu.2020.01861. eCollection 2020. Front Immunol. 2020. PMID: 32973768 Free PMC article.
-
Peptide vaccination directed against IDO1-expressing immune cells elicits CD8+ and CD4+ T-cell-mediated antitumor immunity and enhanced anti-PD1 responses.J Immunother Cancer. 2020 Jul;8(2):e000605. doi: 10.1136/jitc-2020-000605. J Immunother Cancer. 2020. PMID: 32690770 Free PMC article.
-
Tumor indoleamine 2,3-dioxygenase (IDO) inhibits CD19-CAR T cells and is downregulated by lymphodepleting drugs.Blood. 2015 Jun 18;125(25):3905-16. doi: 10.1182/blood-2015-01-621474. Epub 2015 May 4. Blood. 2015. PMID: 25940712 Free PMC article.
-
Insights into Immune Exhaustion in Chronic Hepatitis B: A Review of Checkpoint Receptor Expression.Pharmaceuticals (Basel). 2024 Jul 21;17(7):964. doi: 10.3390/ph17070964. Pharmaceuticals (Basel). 2024. PMID: 39065812 Free PMC article. Review.
-
Trial watch: IDO inhibitors in cancer therapy.Oncoimmunology. 2020 Jun 14;9(1):1777625. doi: 10.1080/2162402X.2020.1777625. Oncoimmunology. 2020. PMID: 32934882 Free PMC article. Review.
References
-
- Prendergast GC, Jaffee EM. Cancer immunologists and cancer biologists: why we didn’t talk then but need to now. Cancer Res. 2007;67(8):3500–3504. - PubMed
-
- Prendergast GC, Jaffee EM, editors. Cancer immunotherapy: immune suppression and tumor growth. 2. New York: Academic Press; 2013.
-
- Muller AJ, Prendergast GC. Marrying immunotherapy with chemotherapy: why say IDO? Cancer Res. 2005;65(18):8065–8068. - PubMed
-
- Muller AJ, DuHadaway JB, Sutanto-Ward E, Donover PS, Prendergast GC. Inhibition of indoleamine 2,3-dioxygenase, an immunomodulatory target of the tumor suppressor gene Bin1, potentiates cancer chemotherapy. Nature Med. 2005;11:312–319. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
