

Innate nuclear sensor IFI16 translocates into the cytoplasm during the early stage of in vitro human cytomegalovirus infection and is entrapped in the egressing virions during the late stage
- PMID: 24696486
- PMCID: PMC4054358
- DOI: 10.1128/JVI.00384-14
Innate nuclear sensor IFI16 translocates into the cytoplasm during the early stage of in vitro human cytomegalovirus infection and is entrapped in the egressing virions during the late stage
Abstract
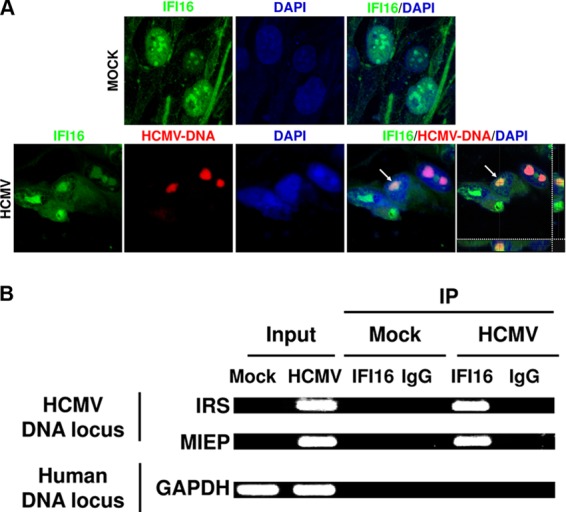
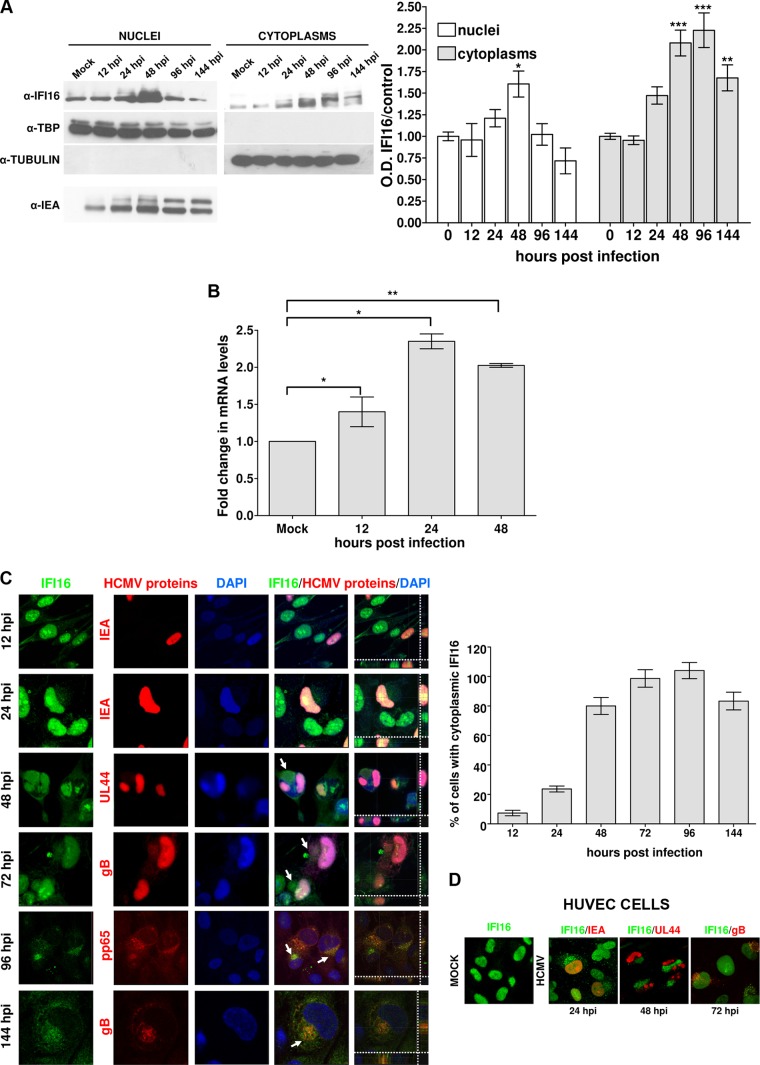
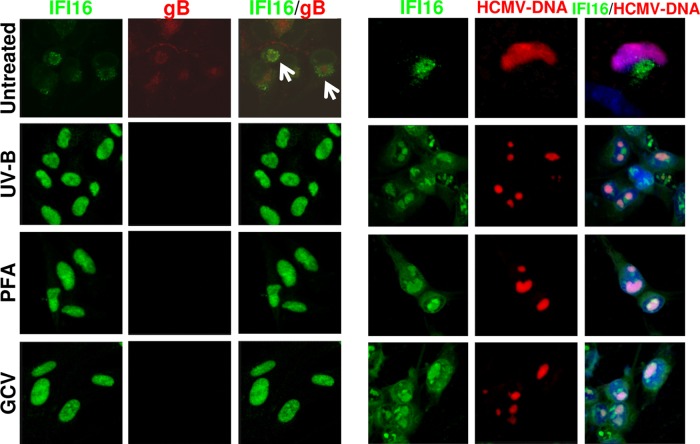
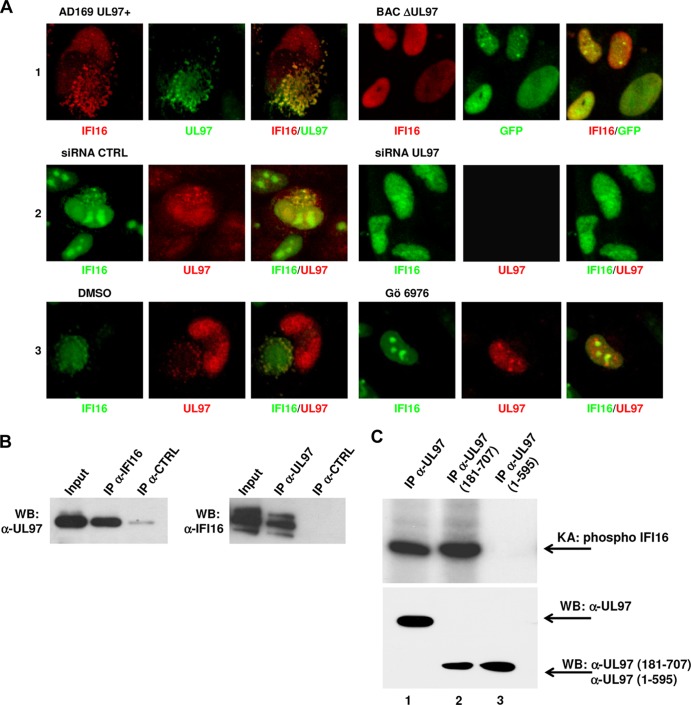
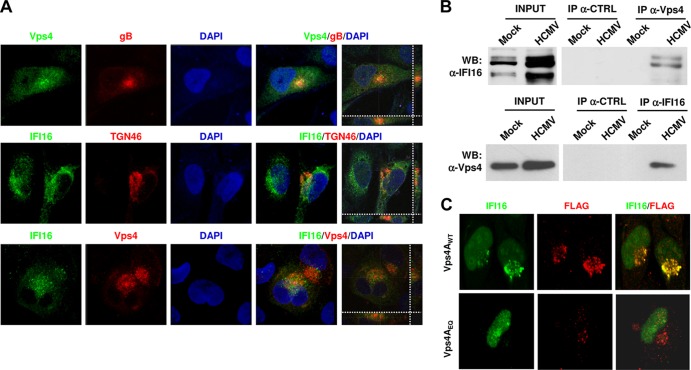
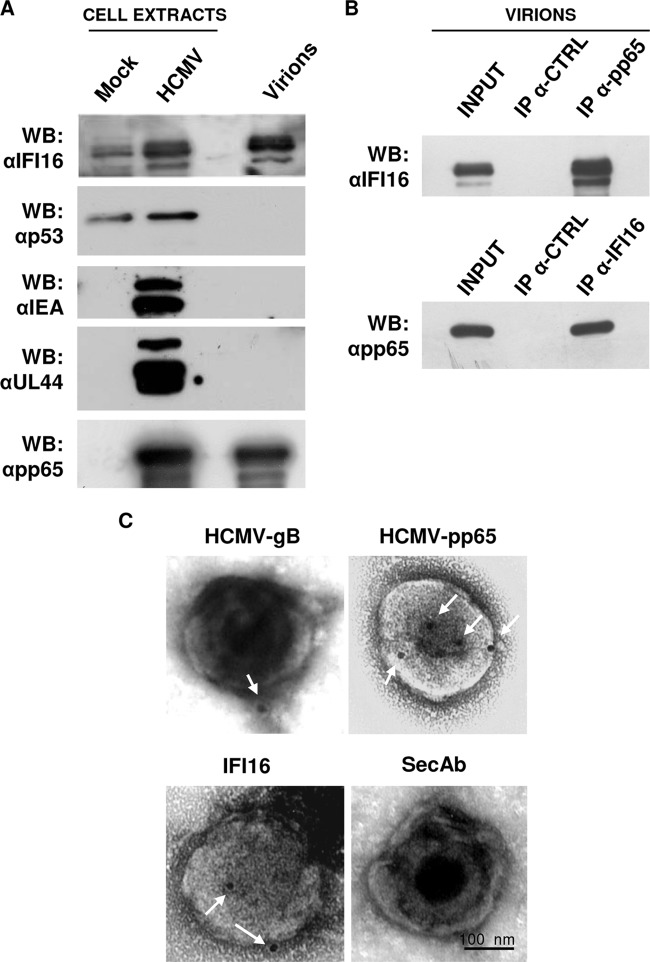
Intrinsic immune mechanisms mediated by constitutively expressed proteins termed "restriction factors" provide frontline antiviral defense. We recently demonstrated that the DNA sensor IFI16 restricts human cytomegalovirus (HCMV) replication by downregulating viral early and late but not immediate-early mRNAs and their protein expression. We show here that at an early time point during the in vitro infection of low-passage-number human embryonic lung fibroblasts, IFI16 binds to HCMV DNA. However, during a later phase following infection, IFI16 is mislocalized to the cytoplasmic virus assembly complex (AC), where it colocalizes with viral structural proteins. Indeed, upon its binding to pUL97, IFI16 undergoes phosphorylation and relocalizes to the cytoplasm of HCMV-infected cells. ESCRT (endosomal sorting complex required for transport) machinery regulates the translocation of IFI16 into the virus AC by sorting and trafficking IFI16 into multivesicular bodies (MVB), as demonstrated by the interaction of IFI16 with two MVB markers: Vps4 and TGN46. Finally, IFI16 becomes incorporated into the newly assembled virions as demonstrated by Western blotting of purified virions and electron microscopy. Together, these results suggest that HCMV has evolved mechanisms to mislocalize and hijack IFI16, trapping it within mature virions. However, the significance of this IFI16 trapping following nuclear mislocalization remains to be established.
Importance: Intracellular viral DNA sensors and restriction factors are critical components of host defense, which alarm and sensitize immune system against intruding pathogens. We have recently demonstrated that the DNA sensor IFI16 restricts human cytomegalovirus (HCMV) replication by downregulating viral early and late but not immediate-early mRNAs and their protein expression. However, viruses are known to evolve numerous strategies to cope and counteract such restriction factors and neutralize the first line of host defense mechanisms. Our findings describe that during early stages of infection, IFI16 successfully recognizes HCMV DNA. However, in late stages HCMV mislocalizes IFI16 into the cytoplasmic viral assembly complex and finally entraps the protein into mature virions. We clarify here the mechanisms HCMV relies to overcome intracellular viral restriction, which provides new insights about the relevance of DNA sensors during HCMV infection.
Copyright © 2014, American Society for Microbiology. All Rights Reserved.
Figures
Similar articles
-
Regulatory Interaction between the Cellular Restriction Factor IFI16 and Viral pp65 (pUL83) Modulates Viral Gene Expression and IFI16 Protein Stability.J Virol. 2016 Aug 26;90(18):8238-50. doi: 10.1128/JVI.00923-16. Print 2016 Sep 15. J Virol. 2016. PMID: 27384655 Free PMC article.
-
The intracellular DNA sensor IFI16 gene acts as restriction factor for human cytomegalovirus replication.PLoS Pathog. 2012 Jan;8(1):e1002498. doi: 10.1371/journal.ppat.1002498. Epub 2012 Jan 26. PLoS Pathog. 2012. PMID: 22291595 Free PMC article.
-
The Nuclear DNA Sensor IFI16 Acts as a Restriction Factor for Human Papillomavirus Replication through Epigenetic Modifications of the Viral Promoters.J Virol. 2015 Aug;89(15):7506-20. doi: 10.1128/JVI.00013-15. Epub 2015 May 13. J Virol. 2015. PMID: 25972554 Free PMC article.
-
Tegument proteins of human cytomegalovirus.Microbiol Mol Biol Rev. 2008 Jun;72(2):249-65, table of contents. doi: 10.1128/MMBR.00040-07. Microbiol Mol Biol Rev. 2008. PMID: 18535146 Free PMC article. Review.
-
Modulation of the innate immune response by human cytomegalovirus.Infect Genet Evol. 2018 Oct;64:105-114. doi: 10.1016/j.meegid.2018.06.025. Epub 2018 Jun 20. Infect Genet Evol. 2018. PMID: 29935337 Review.
Cited by
-
Evasion of Intracellular DNA Sensing by Human Herpesviruses.Front Cell Infect Microbiol. 2021 Mar 15;11:647992. doi: 10.3389/fcimb.2021.647992. eCollection 2021. Front Cell Infect Microbiol. 2021. PMID: 33791247 Free PMC article. Review.
-
The battle between host antiviral innate immunity and immune evasion by cytomegalovirus.Cell Mol Life Sci. 2024 Aug 9;81(1):341. doi: 10.1007/s00018-024-05369-y. Cell Mol Life Sci. 2024. PMID: 39120730 Free PMC article. Review.
-
Emerging Role of PYHIN Proteins as Antiviral Restriction Factors.Viruses. 2020 Dec 18;12(12):1464. doi: 10.3390/v12121464. Viruses. 2020. PMID: 33353088 Free PMC article. Review.
-
The roles of interferon-inducible p200 family members IFI16 and p204 in innate immune responses, cell differentiation and proliferation.Genes Dis. 2015 Mar;2(1):46-56. doi: 10.1016/j.gendis.2014.10.003. Genes Dis. 2015. PMID: 25815367 Free PMC article.
-
DNA double-strand break repair and nucleic acid-related immunity.Acta Biochim Biophys Sin (Shanghai). 2022 May 25;54(6):828-835. doi: 10.3724/abbs.2022061. Acta Biochim Biophys Sin (Shanghai). 2022. PMID: 35975605 Free PMC article. Review.
