

Widespread context dependency of microRNA-mediated regulation
- PMID: 24668909
- PMCID: PMC4032855
- DOI: 10.1101/gr.166702.113
Widespread context dependency of microRNA-mediated regulation
Abstract
Gene expression is regulated in a context-dependent, cell-type-specific manner. Condition-specific transcription is dependent on the presence of transcription factors (TFs) that can activate or inhibit its target genes (global context). Additional factors, such as chromatin structure, histone, or DNA modifications, also influence the activity of individual target genes (individual context). The role of the global and individual context for post-transcriptional regulation has not systematically been investigated on a large scale and is poorly understood. Here we show that global and individual context dependency is a pervasive feature of microRNA-mediated regulation. Our comprehensive and highly consistent data set from several high-throughput technologies (PAR-CLIP, RIP-chip, 4sU-tagging, and SILAC) provides strong evidence that context-dependent microRNA target sites (CDTS) are as frequent and functionally relevant as constitutive target sites (CTS). Furthermore, we found the global context to be insufficient to explain the CDTS, and that flanking sequence motifs provide individual context that is an equally important factor. Our results demonstrate that, similar to TF-mediated regulation, global and individual context dependency are prevalent in microRNA-mediated gene regulation, implying a much more complex post-transcriptional regulatory network than is currently known. The necessary tools to unravel post-transcriptional regulations and mechanisms need to be much more involved, and much more data will be needed for particular cell types and cellular conditions in order to understand microRNA-mediated regulation and the context-dependent post-transcriptional regulatory network.
© 2014 Erhard et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
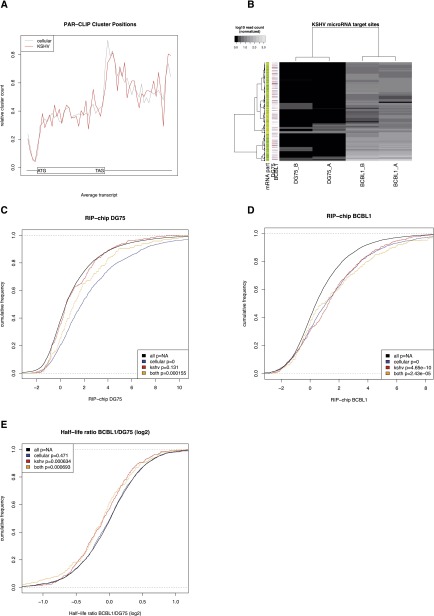
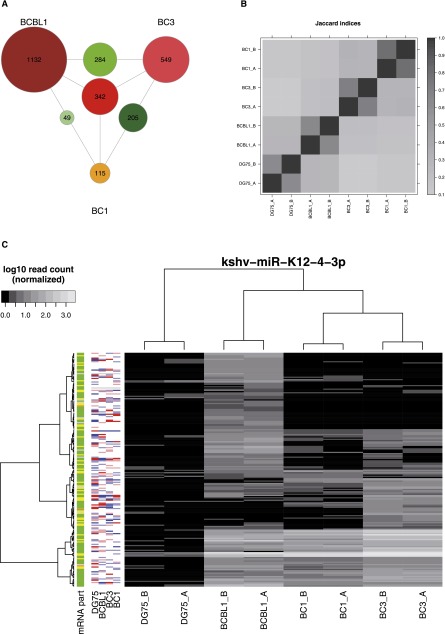
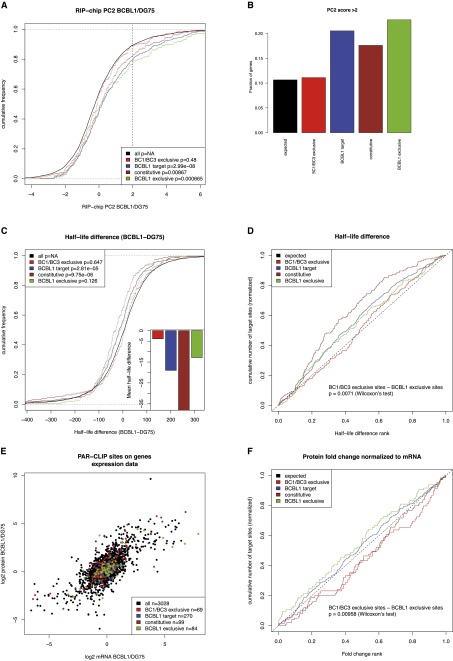
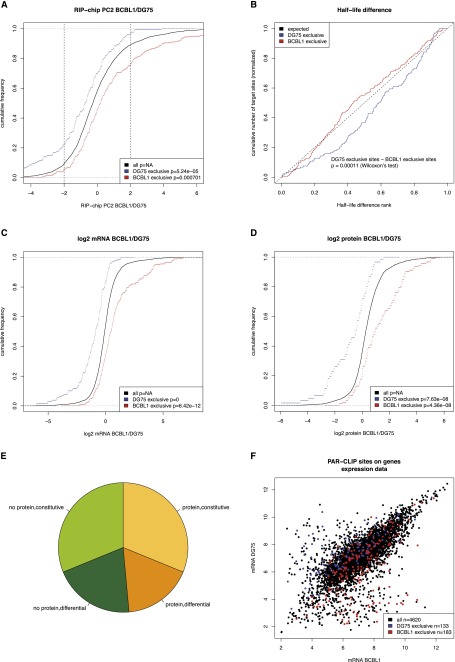
Similar articles
-
Integrated analyses to reconstruct microRNA-mediated regulatory networks in mouse liver using high-throughput profiling.BMC Genomics. 2015;16 Suppl 2(Suppl 2):S12. doi: 10.1186/1471-2164-16-S2-S12. Epub 2015 Jan 21. BMC Genomics. 2015. PMID: 25707768 Free PMC article.
-
Linking Hematopoietic Differentiation to Co-Expressed Sets of Pluripotency-Associated and Imprinted Genes and to Regulatory microRNA-Transcription Factor Motifs.PLoS One. 2017 Jan 4;12(1):e0166852. doi: 10.1371/journal.pone.0166852. eCollection 2017. PLoS One. 2017. PMID: 28052084 Free PMC article.
-
An integrated approach to characterize transcription factor and microRNA regulatory networks involved in Schwann cell response to peripheral nerve injury.BMC Genomics. 2013 Feb 6;14:84. doi: 10.1186/1471-2164-14-84. BMC Genomics. 2013. PMID: 23387820 Free PMC article.
-
A guide to microRNA-mediated gene silencing.FEBS J. 2019 Feb;286(4):642-652. doi: 10.1111/febs.14666. Epub 2018 Oct 8. FEBS J. 2019. PMID: 30267606 Review.
-
From Endogenous to Synthetic microRNA-Mediated Regulatory Circuits: An Overview.Cells. 2019 Nov 29;8(12):1540. doi: 10.3390/cells8121540. Cells. 2019. PMID: 31795372 Free PMC article. Review.
Cited by
-
Identification and functional validation of SRC and RAPGEF1 as new direct targets of miR-203, involved in regulation of epidermal homeostasis.Sci Rep. 2023 Aug 27;13(1):14006. doi: 10.1038/s41598-023-40441-w. Sci Rep. 2023. PMID: 37635193 Free PMC article.
-
Sex-Specific MicroRNAs in Neurovascular Units in Ischemic Stroke.Int J Mol Sci. 2021 Nov 2;22(21):11888. doi: 10.3390/ijms222111888. Int J Mol Sci. 2021. PMID: 34769320 Free PMC article. Review.
-
A systematic approach to RNA-associated motif discovery.BMC Genomics. 2018 Feb 14;19(1):146. doi: 10.1186/s12864-018-4528-x. BMC Genomics. 2018. PMID: 29444662 Free PMC article.
-
miR-762 can negatively regulate menin in ovarian cancer.Onco Targets Ther. 2017 Apr 12;10:2127-2137. doi: 10.2147/OTT.S127872. eCollection 2017. Onco Targets Ther. 2017. PMID: 28442921 Free PMC article.
-
miR-448 suppresses proliferation and invasion by regulating IGF1R in colorectal cancer cells.Am J Transl Res. 2016 Jul 15;8(7):3013-22. eCollection 2016. Am J Transl Res. 2016. Retraction in: Am J Transl Res. 2022 Jan 15;14(1):696. PMID: 27508021 Free PMC article. Retracted.
References
-
- Baltz AG, Munschauer M, Schwanhäusser B, Vasile A, Murakawa Y, Schueler M, Youngs N, Penfold-Brown D, Drew K, Milek M, et al. 2012. The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Mol Cell 46: 674–690 - PubMed
-
- Bartel DP 2004. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116: 281–297 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous