

Host microRNA regulation of human cytomegalovirus immediate early protein translation promotes viral latency
- PMID: 24599990
- PMCID: PMC4019081
- DOI: 10.1128/JVI.00481-14
Host microRNA regulation of human cytomegalovirus immediate early protein translation promotes viral latency
Abstract
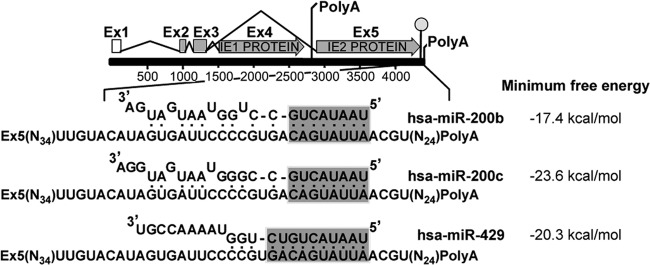
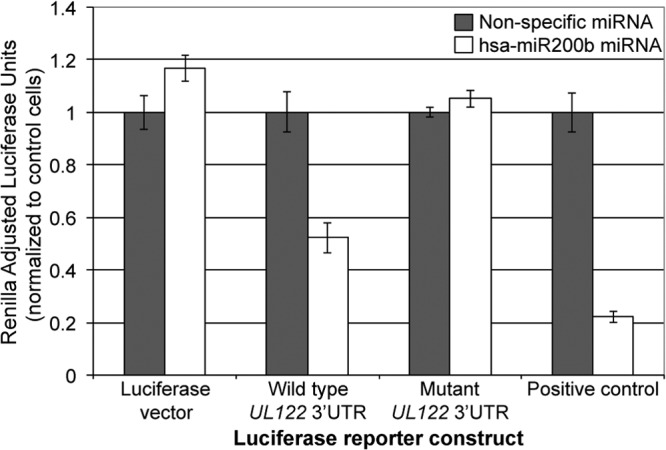
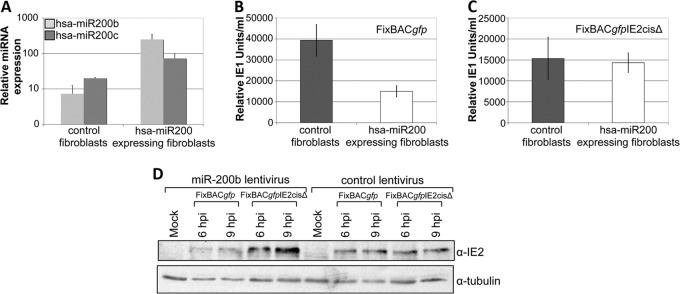
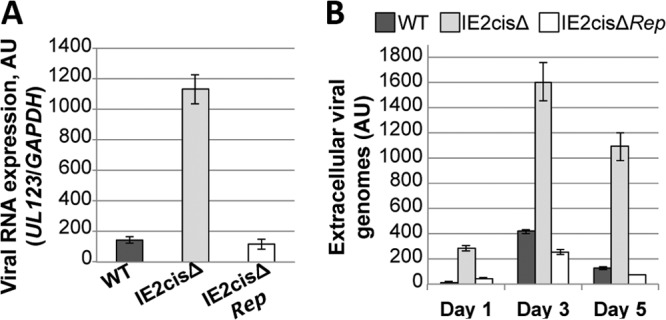
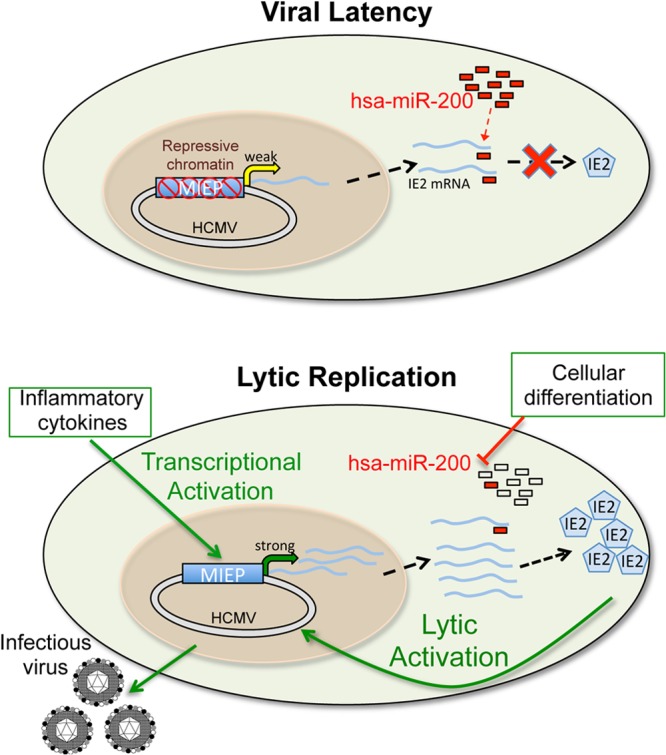
Reactivation of human cytomegalovirus (HCMV) is a significant cause of disease and death in immunocompromised patients, underscoring the need to understand how latency is controlled. Here we demonstrate that HCMV has evolved to utilize cellular microRNAs (miRNAs) in cells that promote latency to regulate expression of a viral protein critical for viral reactivation. Our data reveal that hsa-miR-200 miRNA family members target the UL122 (immediate early protein 2) 3' untranslated region, resulting in repression of this viral protein. Utilizing recombinant viruses that mutate the miRNA-binding site compared to the sequence of the wild-type virus results in lytic rather than latent infections in ex vivo infections of primary CD34+ cells. Cells permissive for lytic replication demonstrate low levels of these miRNAs. We propose that cellular miRNA regulation of HCMV is critical for maintenance of viral latency.
Importance: Human cytomegalovirus (HCMV) is a herpesvirus that infects a majority of the population. Once acquired, individuals harbor the virus for life, where the virus remains, for the most part, in a quiet or latent state. Under weakened immune conditions, the virus can reactivate, which can cause severe disease and often death. We have found that members of a family of small RNAs, termed microRNAs, encoded by human myeloid progenitor cells are capable of repressing a key viral protein, thus enabling the virus to ensure a quiet/latent state. As these progenitor cells mature further down the myeloid lineage toward cells that support active viral replication, the levels of these microRNAs decrease. Together, our data suggest that host cell microRNA regulation of HCMV is important for the quiet/latent state of this pathogen.
Figures
Similar articles
-
Human cytomegalovirus latent infection alters the expression of cellular and viral microRNA.Gene. 2014 Feb 25;536(2):272-8. doi: 10.1016/j.gene.2013.12.012. Epub 2013 Dec 18. Gene. 2014. PMID: 24361963
-
Human Cytomegalovirus US28 Ligand Binding Activity Is Required for Latency in CD34+ Hematopoietic Progenitor Cells and Humanized NSG Mice.mBio. 2019 Aug 20;10(4):e01889-19. doi: 10.1128/mBio.01889-19. mBio. 2019. PMID: 31431555 Free PMC article.
-
Alternative promoters drive human cytomegalovirus reactivation from latency.Proc Natl Acad Sci U S A. 2019 Aug 27;116(35):17492-17497. doi: 10.1073/pnas.1900783116. Epub 2019 Aug 13. Proc Natl Acad Sci U S A. 2019. PMID: 31409717 Free PMC article.
-
Sleepless latency of human cytomegalovirus.Med Microbiol Immunol. 2015 Jun;204(3):421-9. doi: 10.1007/s00430-015-0401-6. Epub 2015 Mar 14. Med Microbiol Immunol. 2015. PMID: 25772624 Free PMC article. Review.
-
Human cytomegalovirus-encoded MicroRNAs: A master regulator of latent infection.Infect Genet Evol. 2020 Mar;78:104119. doi: 10.1016/j.meegid.2019.104119. Epub 2019 Nov 15. Infect Genet Evol. 2020. PMID: 31740397 Review.
Cited by
-
Long and Short Isoforms of the Human Cytomegalovirus UL138 Protein Silence IE Transcription and Promote Latency.J Virol. 2016 Sep 29;90(20):9483-94. doi: 10.1128/JVI.01547-16. Print 2016 Oct 15. J Virol. 2016. PMID: 27512069 Free PMC article.
-
HHV-6 encoded small non-coding RNAs define an intermediate and early stage in viral reactivation.NPJ Genom Med. 2018 Sep 5;3:25. doi: 10.1038/s41525-018-0064-5. eCollection 2018. NPJ Genom Med. 2018. PMID: 30210807 Free PMC article.
-
Host microRNA analysis in cyprinid Herpesvirus-3 (CyHV-3) infected common carp.BMC Genomics. 2019 Jan 16;20(1):46. doi: 10.1186/s12864-018-5266-9. BMC Genomics. 2019. PMID: 30654758 Free PMC article.
-
Human MicroRNAs Attenuate the Expression of Immediate Early Proteins and HCMV Replication during Lytic and Latent Infection in Connection with Enhancement of Phosphorylated RelA/p65 (Serine 536) That Binds to MIEP.Int J Mol Sci. 2022 Mar 2;23(5):2769. doi: 10.3390/ijms23052769. Int J Mol Sci. 2022. PMID: 35269913 Free PMC article.
-
MicroRNA Involvement in Signaling Pathways During Viral Infection.Front Cell Dev Biol. 2020 Mar 10;8:143. doi: 10.3389/fcell.2020.00143. eCollection 2020. Front Cell Dev Biol. 2020. PMID: 32211411 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
