

Antibody-mediated inhibition of TNFR1 attenuates disease in a mouse model of multiple sclerosis
- PMID: 24587232
- PMCID: PMC3938650
- DOI: 10.1371/journal.pone.0090117
Antibody-mediated inhibition of TNFR1 attenuates disease in a mouse model of multiple sclerosis
Abstract
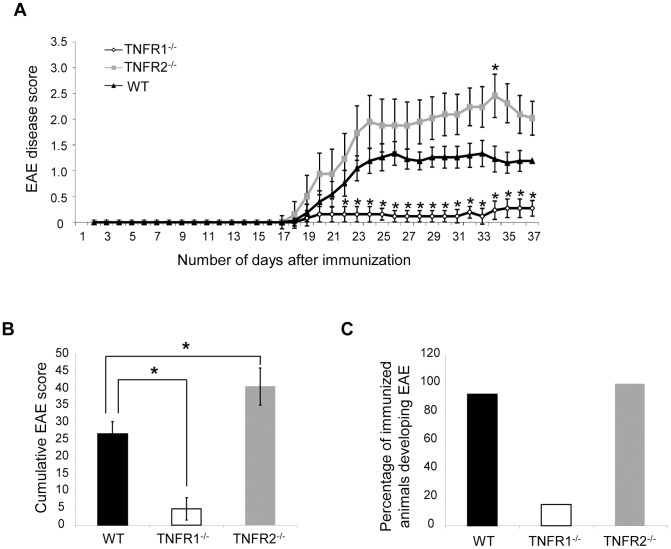
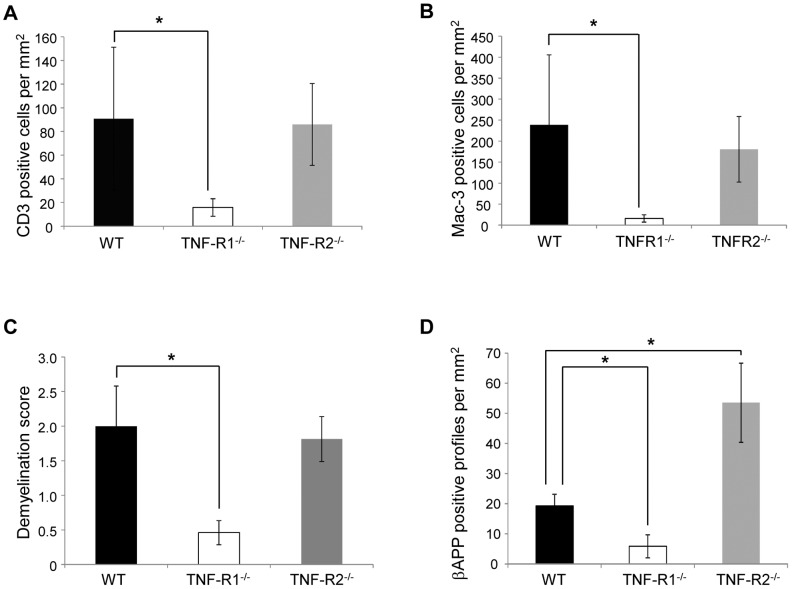
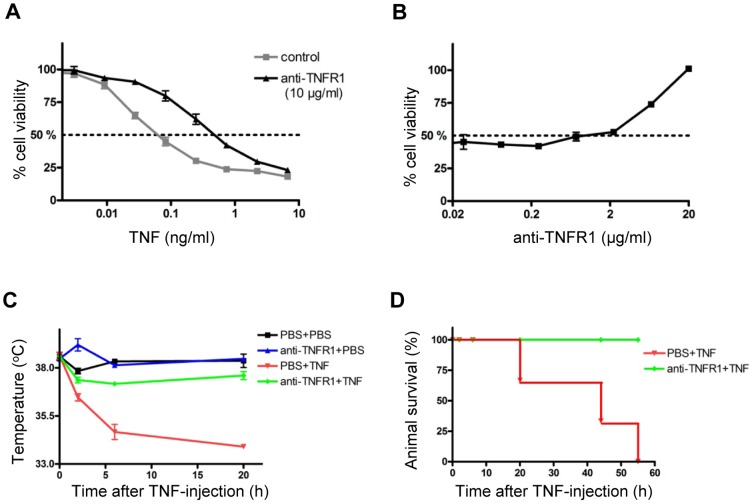
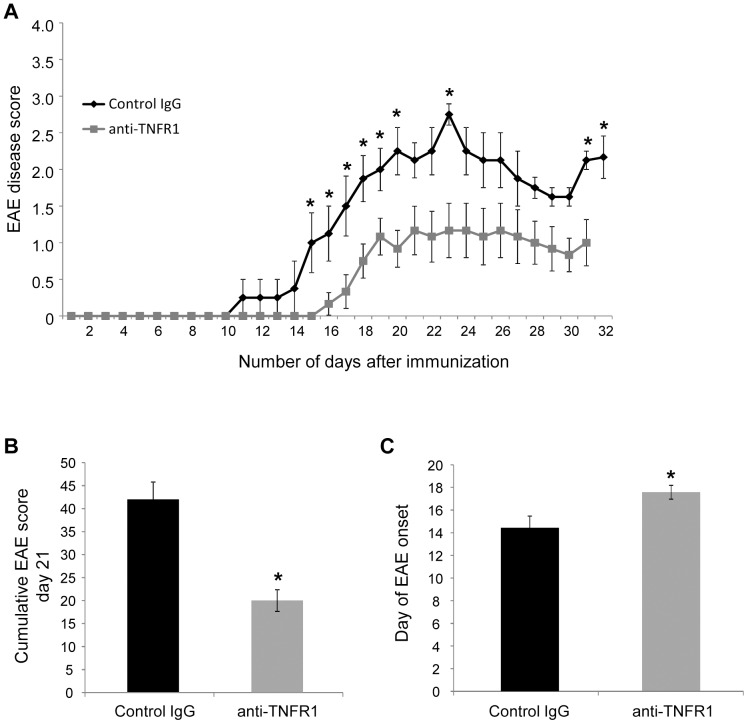
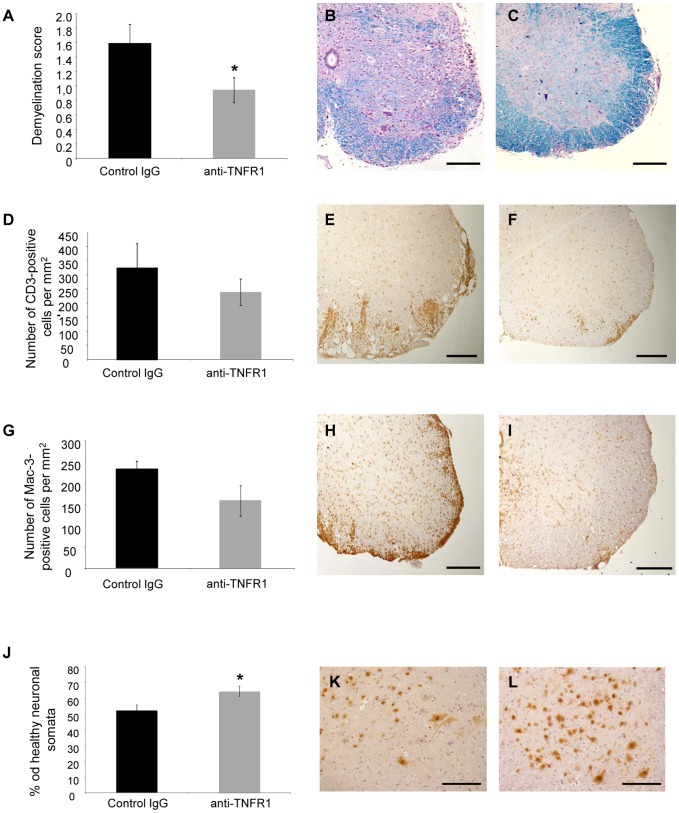
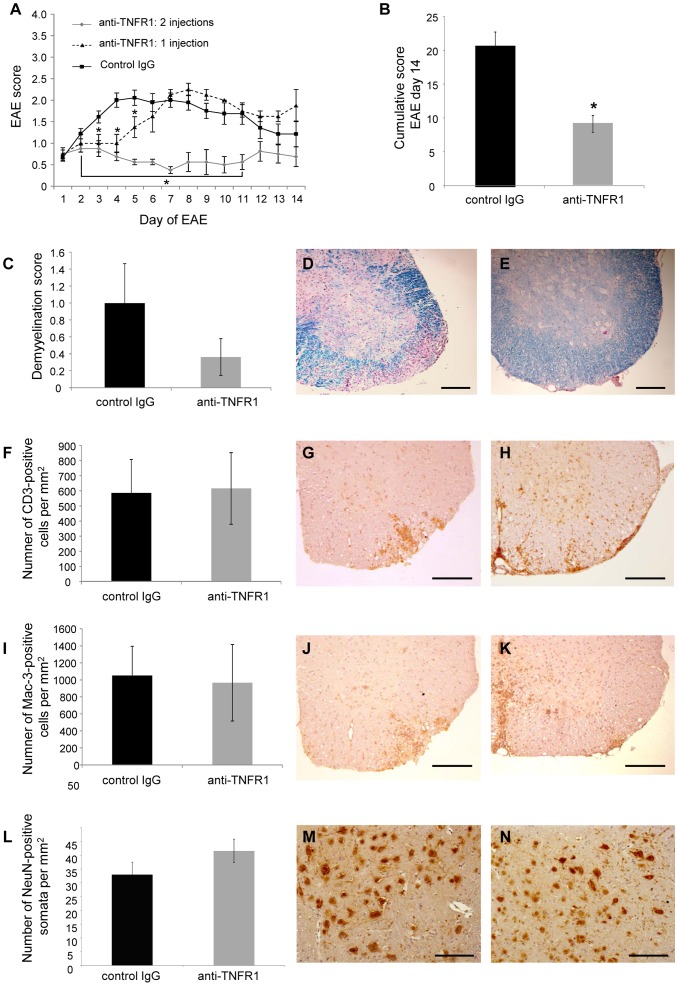
Tumour necrosis factor (TNF) is a proinflammatory cytokine that is known to regulate inflammation in a number of autoimmune diseases, including multiple sclerosis (MS). Although targeting of TNF in models of MS has been successful, the pathological role of TNF in MS remains unclear due to clinical trials where the non-selective inhibition of TNF resulted in exacerbated disease. Subsequent experiments have indicated that this may have resulted from the divergent effects of the two TNF receptors, TNFR1 and TNFR2. Here we show that the selective targeting of TNFR1 with an antagonistic antibody ameliorates symptoms of the most common animal model of MS, experimental autoimmune encephalomyelitis (EAE), when given following both a prophylactic and therapeutic treatment regime. Our results demonstrate that antagonistic TNFR1-specific antibodies may represent a therapeutic approach for the treatment of MS in the future.
Conflict of interest statement
Figures
Similar articles
-
Anti-TNFR1 targeting in humanized mice ameliorates disease in a model of multiple sclerosis.Sci Rep. 2018 Sep 11;8(1):13628. doi: 10.1038/s41598-018-31957-7. Sci Rep. 2018. PMID: 30206422 Free PMC article.
-
Co-modulation of TNFR1 and TNFR2 in an animal model of multiple sclerosis.J Neuroinflammation. 2023 Apr 30;20(1):100. doi: 10.1186/s12974-023-02784-z. J Neuroinflammation. 2023. PMID: 37122019 Free PMC article.
-
TNFR1 inhibition with a Nanobody protects against EAE development in mice.Sci Rep. 2017 Oct 20;7(1):13646. doi: 10.1038/s41598-017-13984-y. Sci Rep. 2017. PMID: 29057962 Free PMC article.
-
Distinct modes of TNF signaling through its two receptors in health and disease.J Leukoc Biol. 2020 Jun;107(6):893-905. doi: 10.1002/JLB.2MR0120-510R. Epub 2020 Feb 21. J Leukoc Biol. 2020. PMID: 32083339 Review.
-
Monoclonal antibody therapy in experimental allergic encephalomyelitis and multiple sclerosis.Immunol Res. 2003;28(1):61-78. doi: 10.1385/IR:28:1:61. Immunol Res. 2003. PMID: 12947225 Review.
Cited by
-
The contribution of tumor necrosis factor to multiple sclerosis: a possible role in progression independent of relapse?J Neuroinflammation. 2024 Aug 21;21(1):209. doi: 10.1186/s12974-024-03193-6. J Neuroinflammation. 2024. PMID: 39169320 Free PMC article. Review.
-
Tumor necrosis factor receptor 2 activation elicits sex-specific effects on cortical myelin proteins and functional recovery in a model of multiple sclerosis.Brain Res Bull. 2024 Feb;207:110885. doi: 10.1016/j.brainresbull.2024.110885. Epub 2024 Jan 20. Brain Res Bull. 2024. PMID: 38246200 Free PMC article.
-
A New Venue of TNF Targeting.Int J Mol Sci. 2018 May 11;19(5):1442. doi: 10.3390/ijms19051442. Int J Mol Sci. 2018. PMID: 29751683 Free PMC article. Review.
-
TNFR2 Deficiency Acts in Concert with Gut Microbiota To Precipitate Spontaneous Sex-Biased Central Nervous System Demyelinating Autoimmune Disease.J Immunol. 2015 Nov 15;195(10):4668-84. doi: 10.4049/jimmunol.1501664. Epub 2015 Oct 16. J Immunol. 2015. PMID: 26475926 Free PMC article.
-
Changes in Cerebrospinal Fluid Balance of TNF and TNF Receptors in Naïve Multiple Sclerosis Patients: Early Involvement in Compartmentalised Intrathecal Inflammation.Cells. 2021 Jul 6;10(7):1712. doi: 10.3390/cells10071712. Cells. 2021. PMID: 34359880 Free PMC article.
References
-
- Davie CA, Barker GJ, Webb S, Tofts PS, Thompson AJ, et al. (1995) Persistent functional deficit in multiple sclerosis and autosomal dominant cerebellar ataxia is associated with axon loss. Brain 118: 1583–1592. - PubMed
-
- Losseff NA, Wang L, Lai HM, Yoo DS, Gawne-Cain ML, et al. (1996) Progressive cerebral atrophy in multiple sclerosis. A serial MRI study. Brain 119: 2009–2019. - PubMed
-
- Trapp BD, Ransohoff R, Rudick R (1999) Axonal pathology in multiple sclerosis: relationship to neurologic disability. Curr Opin Neurol 12: 295–302. - PubMed
-
- Wajant H, Pfizenmaier K, Scheurich P (2003) Tumor necrosis factor signaling. Cell Death Diff 10: 45–65. - PubMed
-
- Kontermann RE, Scheurich P, Pfizenmaier K (2009) Antagonists of TNF action: clinical experience and new developments. Expert Opin. Drug Discov 4: 279–292. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
