

Follistatin in chondrocytes: the link between TRPV4 channelopathies and skeletal malformations
- PMID: 24577120
- PMCID: PMC4021446
- DOI: 10.1096/fj.13-245936
Follistatin in chondrocytes: the link between TRPV4 channelopathies and skeletal malformations
Abstract
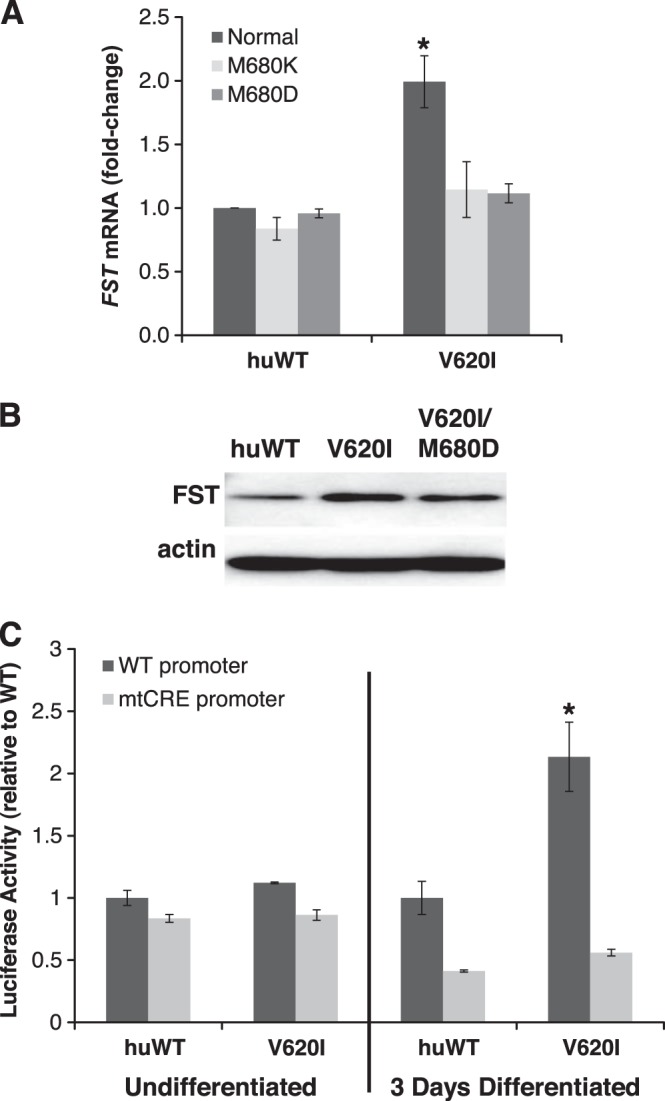
Point mutations in the calcium-permeable TRPV4 ion channel have been identified as the cause of autosomal-dominant human motor neuropathies, arthropathies, and skeletal malformations of varying severity. The objective of this study was to determine the mechanism by which TRPV4 channelopathy mutations cause skeletal dysplasia. The human TRPV4(V620I) channelopathy mutation was transfected into primary porcine chondrocytes and caused significant (2.6-fold) up-regulation of follistatin (FST) expression levels. Pore altering mutations that prevent calcium influx through the channel prevented significant FST up-regulation (1.1-fold). We generated a mouse model of the TRPV4(V620I) mutation, and found significant skeletal deformities (e.g., shortening of tibiae and digits, similar to the human disease brachyolmia) and increases in Fst/TRPV4 mRNA levels (2.8-fold). FST was significantly up-regulated in primary chondrocytes transfected with 3 different dysplasia-causing TRPV4 mutations (2- to 2.3-fold), but was not affected by an arthropathy mutation (1.1-fold). Furthermore, FST-loaded microbeads decreased bone ossification in developing chick femora (6%) and tibiae (11%). FST gene and protein levels were also increased 4-fold in human chondrocytes from an individual natively expressing the TRPV4(T89I) mutation. Taken together, these data strongly support that up-regulation of FST in chondrocytes by skeletal dysplasia-inducing TRPV4 mutations contributes to disease pathogenesis.
Keywords: bone morphogenetic protein; calcium signaling; cartilage; growth plate.
© FASEB.
Figures





Similar articles
-
Skeletal dysplasia-causing TRPV4 mutations suppress the hypertrophic differentiation of human iPSC-derived chondrocytes.Elife. 2023 Feb 22;12:e71154. doi: 10.7554/eLife.71154. Elife. 2023. PMID: 36810131 Free PMC article.
-
Mice expressing mutant Trpv4 recapitulate the human TRPV4 disorders.J Bone Miner Res. 2014 Aug;29(8):1815-1822. doi: 10.1002/jbmr.2220. J Bone Miner Res. 2014. PMID: 24644033 Free PMC article.
-
A mutation in TRPV4 results in altered chondrocyte calcium signaling in severe metatropic dysplasia.Am J Med Genet A. 2015 Oct;167A(10):2286-93. doi: 10.1002/ajmg.a.37182. Epub 2015 Aug 6. Am J Med Genet A. 2015. PMID: 26249260
-
Human skeletal dysplasia caused by a constitutive activated transient receptor potential vanilloid 4 (TRPV4) cation channel mutation.Exp Mol Med. 2012 Dec 31;44(12):707-22. doi: 10.3858/emm.2012.44.12.080. Exp Mol Med. 2012. PMID: 23143559 Free PMC article. Review.
-
TRPV4-pathy, a novel channelopathy affecting diverse systems.J Hum Genet. 2010 Jul;55(7):400-2. doi: 10.1038/jhg.2010.37. Epub 2010 May 27. J Hum Genet. 2010. PMID: 20505684 Review.
Cited by
-
Cartilage-Specific Knockout of the Mechanosensory Ion Channel TRPV4 Decreases Age-Related Osteoarthritis.Sci Rep. 2016 Jul 8;6:29053. doi: 10.1038/srep29053. Sci Rep. 2016. PMID: 27388701 Free PMC article.
-
New therapeutic targets in rare genetic skeletal diseases.Expert Opin Orphan Drugs. 2015 Oct 3;3(10):1137-1154. doi: 10.1517/21678707.2015.1083853. Epub 2015 Sep 24. Expert Opin Orphan Drugs. 2015. PMID: 26635999 Free PMC article.
-
Pleiotropic function of TRPV4 ion channels in the central nervous system.Exp Physiol. 2016 Dec 1;101(12):1472-1476. doi: 10.1113/EP085790. Epub 2016 Nov 8. Exp Physiol. 2016. PMID: 27701788 Free PMC article. Review.
-
Cell Responsiveness to Physical Energies: Paving the Way to Decipher a Morphogenetic Code.Int J Mol Sci. 2022 Mar 15;23(6):3157. doi: 10.3390/ijms23063157. Int J Mol Sci. 2022. PMID: 35328576 Free PMC article. Review.
-
Synergy between Piezo1 and Piezo2 channels confers high-strain mechanosensitivity to articular cartilage.Proc Natl Acad Sci U S A. 2014 Nov 25;111(47):E5114-22. doi: 10.1073/pnas.1414298111. Epub 2014 Nov 10. Proc Natl Acad Sci U S A. 2014. PMID: 25385580 Free PMC article.
References
-
- Nilius B., Voets T. (2004) Diversity of TRP channel activation. Novartis Found. Symp. 258, 140–149; discussion 149–159, 263–266 - PubMed
-
- Liedtke W. (2006) TRPV channels' function in sensory transduction and cellular signaling cascades. In TRP Ion Channel Function in Sensory Transduction and Cellular Signaling Cascades (Liedtke W., Heller S., eds) pp. 303–318, CRC Press/Taylor & Francis, Boca Raton, FL, USA - PubMed
-
- Montell C. (2001) Physiology, phylogeny, and functions of the TRP superfamily of cation channels. Sci. STKE. 2001, RE 1 - PubMed
-
- Nilius B., Watanabe H., Vriens J. (2003) The TRPV4 channel: structure-function relationship and promiscuous gating behaviour. Pflügers Arch. 446, 298–303 - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
- AG15768/AG/NIA NIH HHS/United States
- HD022657/HD/NICHD NIH HHS/United States
- R01 AR048852/AR/NIAMS NIH HHS/United States
- R01 AG046927/AG/NIA NIH HHS/United States
- AG46927/AG/NIA NIH HHS/United States
- 5P30NS061789/NS/NINDS NIH HHS/United States
- P30 NS061789/NS/NINDS NIH HHS/United States
- AR48852/AR/NIAMS NIH HHS/United States
- R01 AR062651/AR/NIAMS NIH HHS/United States
- AR062651/AR/NIAMS NIH HHS/United States
- P01 HD022657/HD/NICHD NIH HHS/United States
- R01 AR048182/AR/NIAMS NIH HHS/United States
- AR50245/AR/NIAMS NIH HHS/United States
- P01 AR050245/AR/NIAMS NIH HHS/United States
- R01 AG015768/AG/NIA NIH HHS/United States
- DE018549/DE/NIDCR NIH HHS/United States
- P41 RR011823/RR/NCRR NIH HHS/United States
- AR48182/AR/NIAMS NIH HHS/United States
- P41RR11823-03/RR/NCRR NIH HHS/United States
- R01 DE018549/DE/NIDCR NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
