

Mutations in RSPH1 cause primary ciliary dyskinesia with a unique clinical and ciliary phenotype
- PMID: 24568568
- PMCID: PMC3983840
- DOI: 10.1164/rccm.201311-2047OC
Mutations in RSPH1 cause primary ciliary dyskinesia with a unique clinical and ciliary phenotype
Abstract
Rationale: Primary ciliary dyskinesia (PCD) is a genetically heterogeneous recessive disorder of motile cilia, but the genetic cause is not defined for all patients with PCD.
Objectives: To identify disease-causing mutations in novel genes, we performed exome sequencing, follow-up characterization, mutation scanning, and genotype-phenotype studies in patients with PCD.
Methods: Whole-exome sequencing was performed using NimbleGen capture and Illumina HiSeq sequencing. Sanger-based sequencing was used for mutation scanning, validation, and segregation analysis.
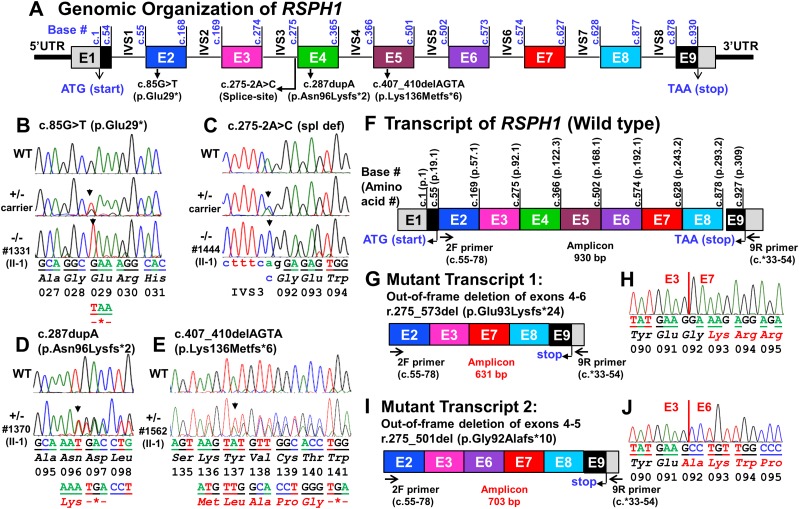
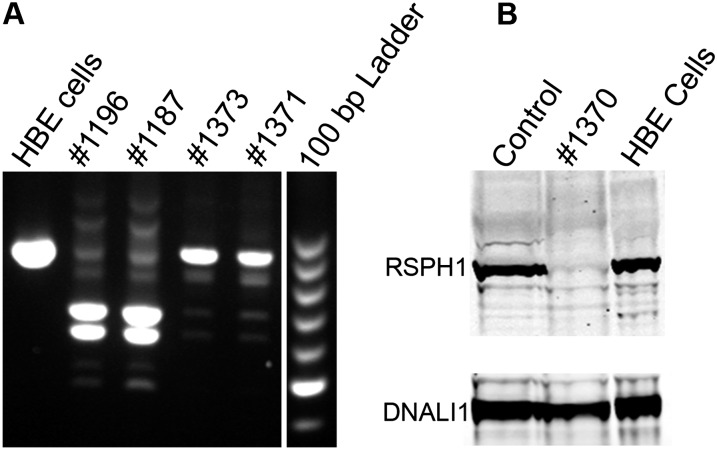
Measurements and main results: We performed exome sequencing on an affected sib-pair with normal ultrastructure in more than 85% of cilia. A homozygous splice-site mutation was detected in RSPH1 in both siblings; parents were carriers. Screening RSPH1 in 413 unrelated probands, including 325 with PCD and 88 with idiopathic bronchiectasis, revealed biallelic loss-of-function mutations in nine additional probands. Five affected siblings of probands in RSPH1 families harbored the familial mutations. The 16 individuals with RSPH1 mutations had some features of PCD; however, nasal nitric oxide levels were higher than in patients with PCD with other gene mutations (98.3 vs. 20.7 nl/min; P < 0.0003). Additionally, individuals with RSPH1 mutations had a lower prevalence (8 of 16) of neonatal respiratory distress, and later onset of daily wet cough than typical for PCD, and better lung function (FEV1), compared with 75 age- and sex-matched PCD cases (73.0 vs. 61.8, FEV1 % predicted; P = 0.043). Cilia from individuals with RSPH1 mutations had normal beat frequency (6.1 ± Hz at 25°C), but an abnormal, circular beat pattern.
Conclusions: The milder clinical disease and higher nasal nitric oxide in individuals with biallelic mutations in RSPH1 provides evidence of a unique genotype-phenotype relationship in PCD, and suggests that mutations in RSPH1 may be associated with residual ciliary function.
Figures



Similar articles
-
Mice with a Deletion of Rsph1 Exhibit a Low Level of Mucociliary Clearance and Develop a Primary Ciliary Dyskinesia Phenotype.Am J Respir Cell Mol Biol. 2019 Sep;61(3):312-321. doi: 10.1165/rcmb.2017-0387OC. Am J Respir Cell Mol Biol. 2019. PMID: 30896965 Free PMC article.
-
Loss-of-function mutations in RSPH1 cause primary ciliary dyskinesia with central-complex and radial-spoke defects.Am J Hum Genet. 2013 Sep 5;93(3):561-70. doi: 10.1016/j.ajhg.2013.07.013. Epub 2013 Aug 29. Am J Hum Genet. 2013. PMID: 23993197 Free PMC article.
-
Targeted NGS gene panel identifies mutations in RSPH1 causing primary ciliary dyskinesia and a common mechanism for ciliary central pair agenesis due to radial spoke defects.Hum Mol Genet. 2014 Jul 1;23(13):3362-74. doi: 10.1093/hmg/ddu046. Epub 2014 Feb 11. Hum Mol Genet. 2014. PMID: 24518672 Free PMC article.
-
[Cilia ultrastructural and gene variation of primary ciliary dyskinesia: report of three cases and literatures review].Zhonghua Er Ke Za Zhi. 2018 Feb 2;56(2):134-137. doi: 10.3760/cma.j.issn.0578-1310.2018.02.012. Zhonghua Er Ke Za Zhi. 2018. PMID: 29429202 Review. Chinese.
-
Value of transmission electron microscopy for primary ciliary dyskinesia diagnosis in the era of molecular medicine: Genetic defects with normal and non-diagnostic ciliary ultrastructure.Ultrastruct Pathol. 2017 Nov-Dec;41(6):373-385. doi: 10.1080/01913123.2017.1362088. Epub 2017 Sep 15. Ultrastruct Pathol. 2017. PMID: 28915070 Free PMC article. Review.
Cited by
-
Subtyping children with asthma by clustering analysis of mRNA expression data.Front Genet. 2022 Sep 9;13:974936. doi: 10.3389/fgene.2022.974936. eCollection 2022. Front Genet. 2022. PMID: 36159986 Free PMC article.
-
Expression of a Truncated Form of ODAD1 Associated with an Unusually Mild Primary Ciliary Dyskinesia Phenotype.Int J Mol Sci. 2022 Feb 3;23(3):1753. doi: 10.3390/ijms23031753. Int J Mol Sci. 2022. PMID: 35163670 Free PMC article.
-
Lower airway clinical outcome measures for use in primary ciliary dyskinesia research: a scoping review.ERJ Open Res. 2021 Nov 29;7(4):00320-2021. doi: 10.1183/23120541.00320-2021. eCollection 2021 Oct. ERJ Open Res. 2021. PMID: 34853782 Free PMC article.
-
Mutations in DNAJB13, Encoding an HSP40 Family Member, Cause Primary Ciliary Dyskinesia and Male Infertility.Am J Hum Genet. 2016 Aug 4;99(2):489-500. doi: 10.1016/j.ajhg.2016.06.022. Am J Hum Genet. 2016. PMID: 27486783 Free PMC article.
-
Primary ciliary dyskinesia caused by a large homozygous deletion including exons 1-4 of DRC1 in Japanese patients with recurrent sinopulmonary infection.Mol Genet Genomic Med. 2020 Jan;8(1):e1033. doi: 10.1002/mgg3.1033. Epub 2019 Nov 8. Mol Genet Genomic Med. 2020. PMID: 31701675 Free PMC article.
References
-
- Zariwala MA, Knowles MR, Omran H. Genetic defects in ciliary structure and function. Annu Rev Physiol. 2007;69:423–450. - PubMed
-
- Bartoloni L, Blouin JL, Pan Y, Gehrig C, Maiti AK, Scamuffa N, Rossier C, Jorissen M, Armengot M, Meeks M, et al. Mutations in the DNAH11 (axonemal heavy chain dynein type 11) gene cause one form of situs inversus totalis and most likely primary ciliary dyskinesia. Proc Natl Acad Sci USA. 2002;99:10282–10286. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- HHSN268201100037C/HL/NHLBI NIH HHS/United States
- UC2 HL103010/HL/NHLBI NIH HHS/United States
- UL1TR000083/TR/NCATS NIH HHS/United States
- 1R01HL117836/HL/NHLBI NIH HHS/United States
- U54 HL096458/HL/NHLBI NIH HHS/United States
- R01 HL117836/HL/NHLBI NIH HHS/United States
- RC2 HL102924/HL/NHLBI NIH HHS/United States
- R01 DK068306/DK/NIDDK NIH HHS/United States
- R01 HL071798/HL/NHLBI NIH HHS/United States
- UM1 HG006493/HG/NHGRI NIH HHS/United States
- RC2 HL103010/HL/NHLBI NIH HHS/United States
- P30 DK065988/DK/NIDDK NIH HHS/United States
- UL1TR000154/TR/NCATS NIH HHS/United States
- RC2 HL102923/HL/NHLBI NIH HHS/United States
- U54 HG006493/HG/NHGRI NIH HHS/United States
- UC2 HL102926/HL/NHLBI NIH HHS/United States
- 5R01HL071798/HL/NHLBI NIH HHS/United States
- RC2 HL102926/HL/NHLBI NIH HHS/United States
- P30-DK065988/DK/NIDDK NIH HHS/United States
- 5 U54HL096458-06/HL/NHLBI NIH HHS/United States
- UL1 TR000154/TR/NCATS NIH HHS/United States
- UC2 HL102923/HL/NHLBI NIH HHS/United States
- UC2 HL102924/HL/NHLBI NIH HHS/United States
- 1 U54HG006493/HG/NHGRI NIH HHS/United States
- UL1 TR000083/TR/NCATS NIH HHS/United States
- UL1 TR001082/TR/NCATS NIH HHS/United States
- RC2 HL102925/HL/NHLBI NIH HHS/United States
- UC2 HL102925/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
