

Interferon-stimulated genes: a complex web of host defenses
- PMID: 24555472
- PMCID: PMC4313732
- DOI: 10.1146/annurev-immunol-032713-120231
Interferon-stimulated genes: a complex web of host defenses
Abstract
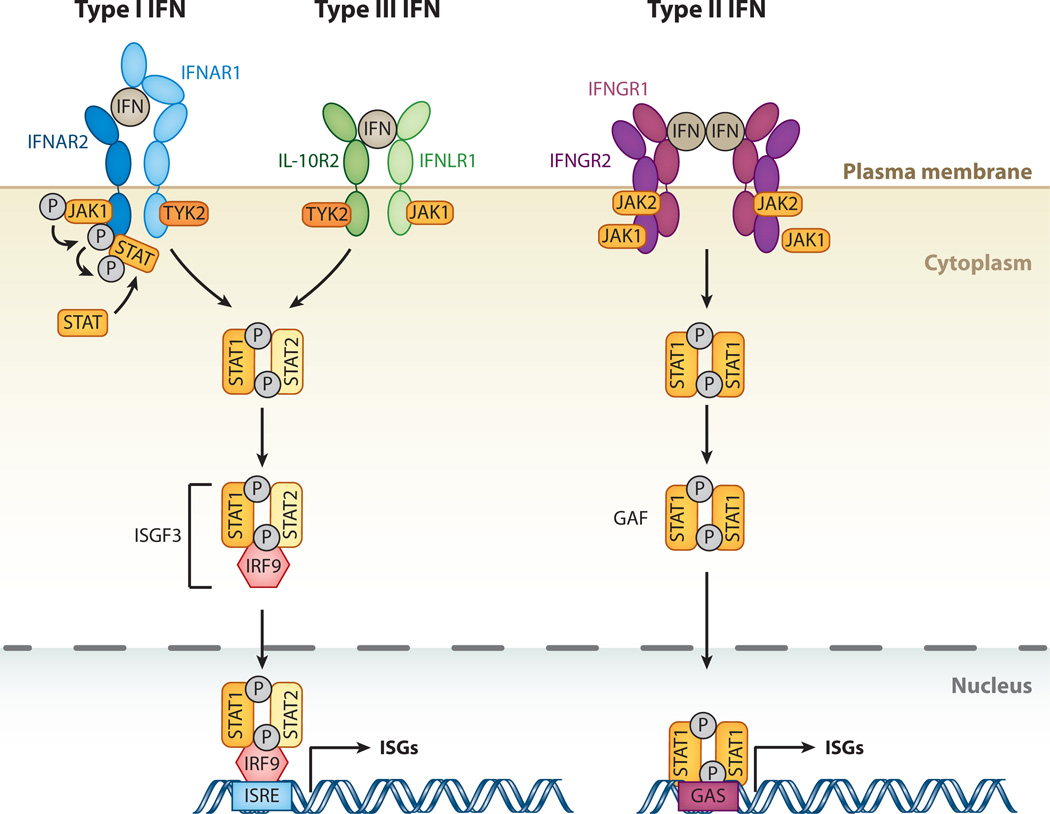
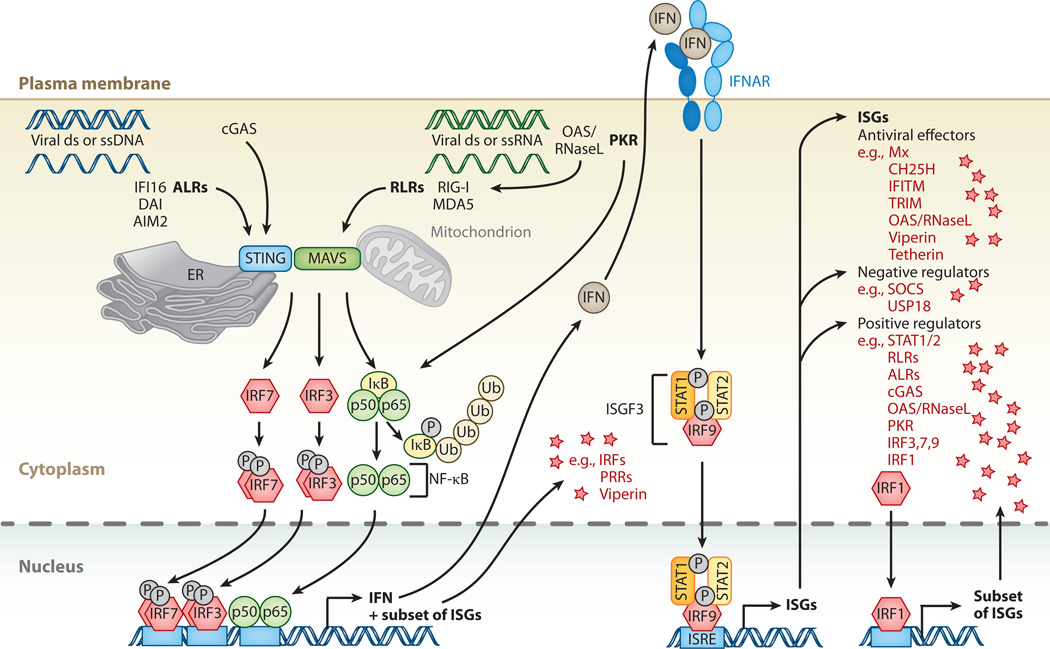
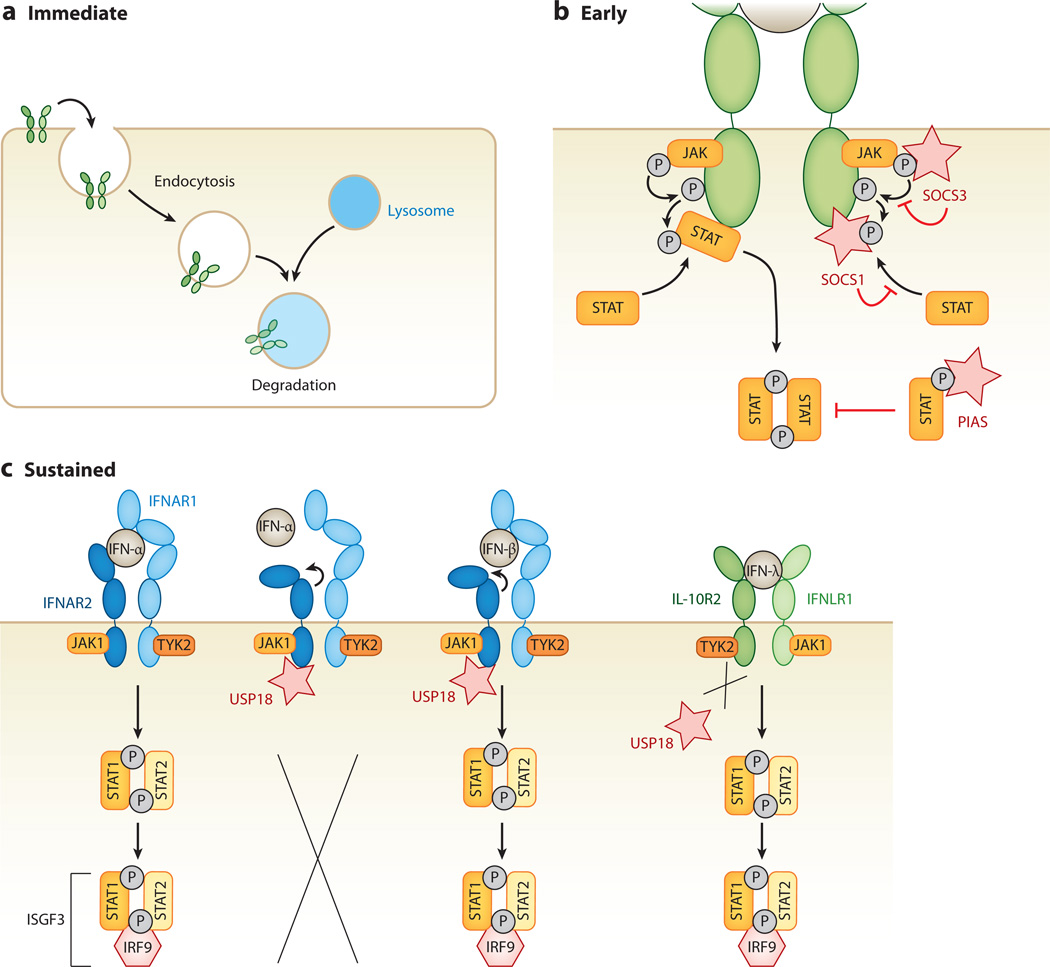
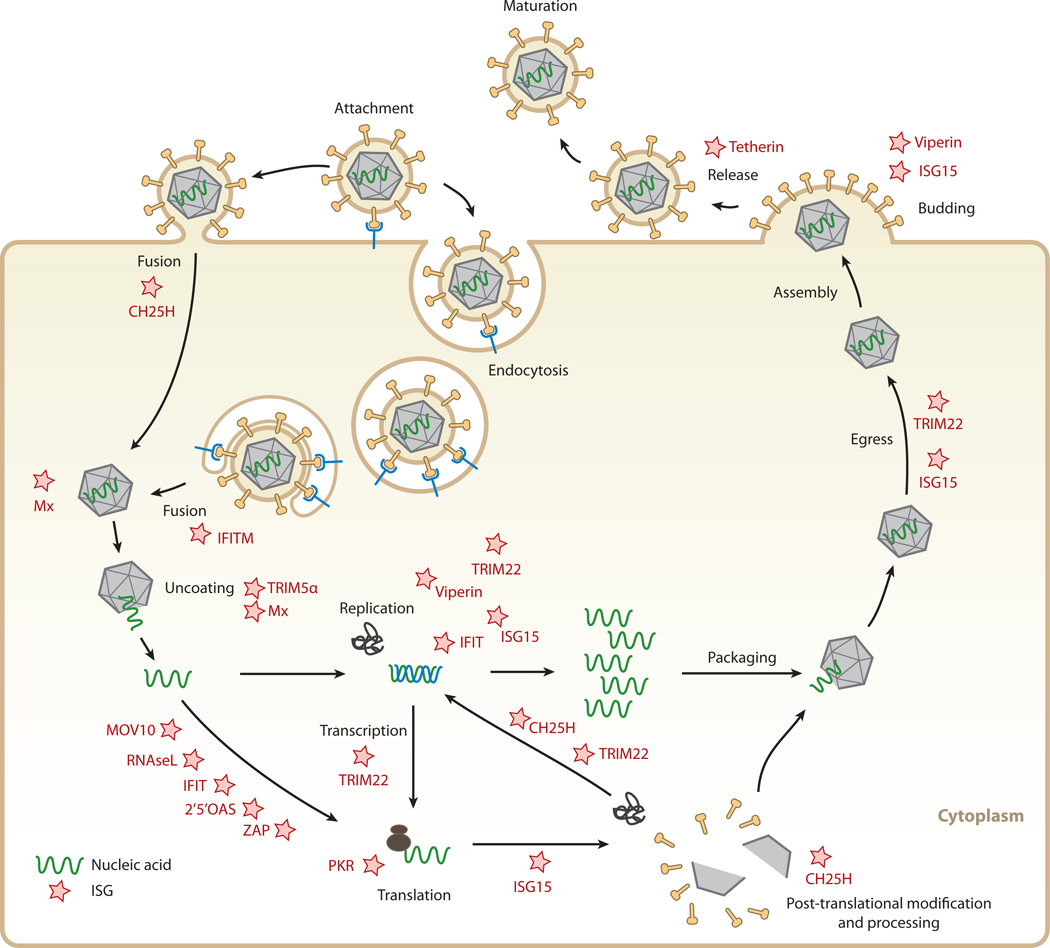
Interferon-stimulated gene (ISG) products take on a number of diverse roles. Collectively, they are highly effective at resisting and controlling pathogens. In this review, we begin by introducing interferon (IFN) and the JAK-STAT signaling pathway to highlight features that impact ISG production. Next, we describe ways in which ISGs both enhance innate pathogen-sensing capabilities and negatively regulate signaling through the JAK-STAT pathway. Several ISGs that directly inhibit virus infection are described with an emphasis on those that impact early and late stages of the virus life cycle. Finally, we describe ongoing efforts to identify and characterize antiviral ISGs, and we provide a forward-looking perspective on the ISG landscape.
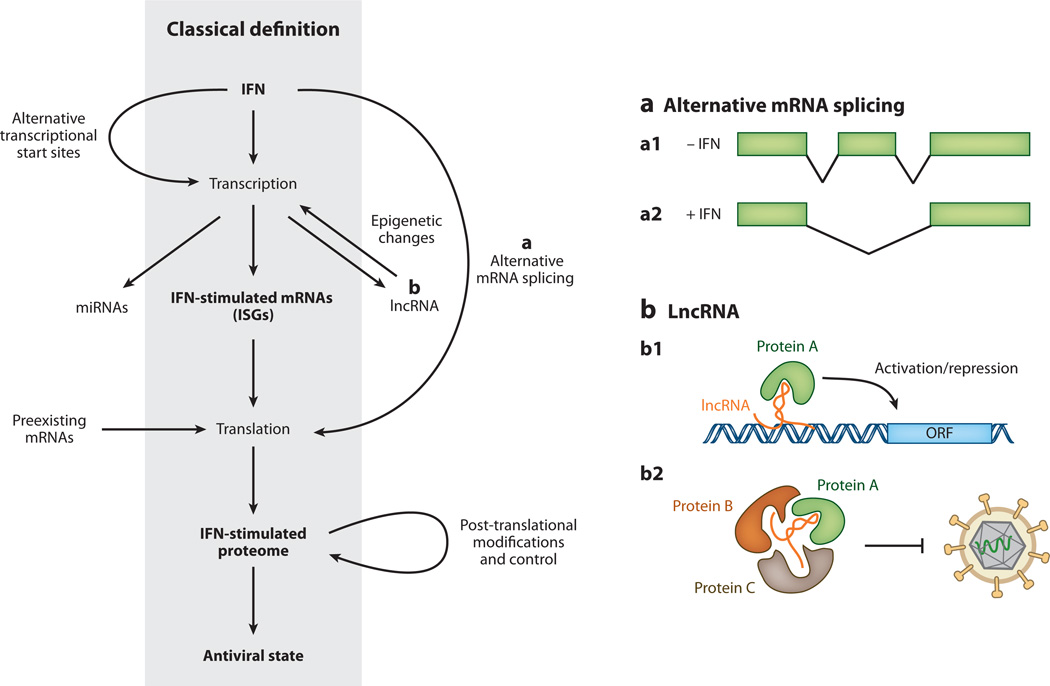
Figures
Similar articles
-
Interference without interferon: interferon-independent induction of interferon-stimulated genes and its role in cellular innate immunity.mBio. 2024 Oct 16;15(10):e0258224. doi: 10.1128/mbio.02582-24. Epub 2024 Sep 20. mBio. 2024. PMID: 39302126 Free PMC article. Review.
-
Immune Cell Profiling of IFN-λ Response Shows pDCs Express Highest Level of IFN-λR1 and Are Directly Responsive via the JAK-STAT Pathway.J Interferon Cytokine Res. 2016 Dec;36(12):671-680. doi: 10.1089/jir.2015.0169. Epub 2016 Sep 12. J Interferon Cytokine Res. 2016. PMID: 27617757 Free PMC article.
-
Herpesviruses: interfering innate immunity by targeting viral sensing and interferon pathways.Rev Med Virol. 2015 May;25(3):187-201. doi: 10.1002/rmv.1836. Epub 2015 Apr 1. Rev Med Virol. 2015. PMID: 25847408 Review.
-
Orf virus inhibits interferon stimulated gene expression and modulates the JAK/STAT signalling pathway.Virus Res. 2015 Oct 2;208:180-8. doi: 10.1016/j.virusres.2015.06.014. Epub 2015 Jun 22. Virus Res. 2015. PMID: 26113305
-
Advances in anti-viral immune defence: revealing the importance of the IFN JAK/STAT pathway.Cell Mol Life Sci. 2017 Jul;74(14):2525-2535. doi: 10.1007/s00018-017-2520-2. Epub 2017 Apr 21. Cell Mol Life Sci. 2017. PMID: 28432378 Free PMC article. Review.
Cited by
-
Organoid modeling of Zika and herpes simplex virus 1 infections reveals virus-specific responses leading to microcephaly.Cell Stem Cell. 2021 Aug 5;28(8):1362-1379.e7. doi: 10.1016/j.stem.2021.03.004. Epub 2021 Apr 9. Cell Stem Cell. 2021. PMID: 33838105 Free PMC article.
-
Human TMEFF1 is a restriction factor for herpes simplex virus in the brain.Nature. 2024 Aug;632(8024):390-400. doi: 10.1038/s41586-024-07745-x. Epub 2024 Jul 24. Nature. 2024. PMID: 39048830 Free PMC article.
-
Flaviviral NS4b, chameleon and jack-in-the-box roles in viral replication and pathogenesis, and a molecular target for antiviral intervention.Rev Med Virol. 2015 Jul;25(4):205-23. doi: 10.1002/rmv.1835. Epub 2015 Apr 1. Rev Med Virol. 2015. PMID: 25828437 Free PMC article. Review.
-
Comprehensive single cell analysis of pandemic influenza A virus infection in the human airways uncovers cell-type specific host transcriptional signatures relevant for disease progression and pathogenesis.Front Immunol. 2022 Oct 4;13:978824. doi: 10.3389/fimmu.2022.978824. eCollection 2022. Front Immunol. 2022. PMID: 36268025 Free PMC article.
-
Expression and mechanisms of interferon-stimulated genes in viral infection of the central nervous system (CNS) and neurological diseases.Front Immunol. 2022 Oct 17;13:1008072. doi: 10.3389/fimmu.2022.1008072. eCollection 2022. Front Immunol. 2022. PMID: 36325336 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
