

Spread, circulation, and evolution of the Middle East respiratory syndrome coronavirus
- PMID: 24549846
- PMCID: PMC3944817
- DOI: 10.1128/mBio.01062-13
Spread, circulation, and evolution of the Middle East respiratory syndrome coronavirus
Abstract
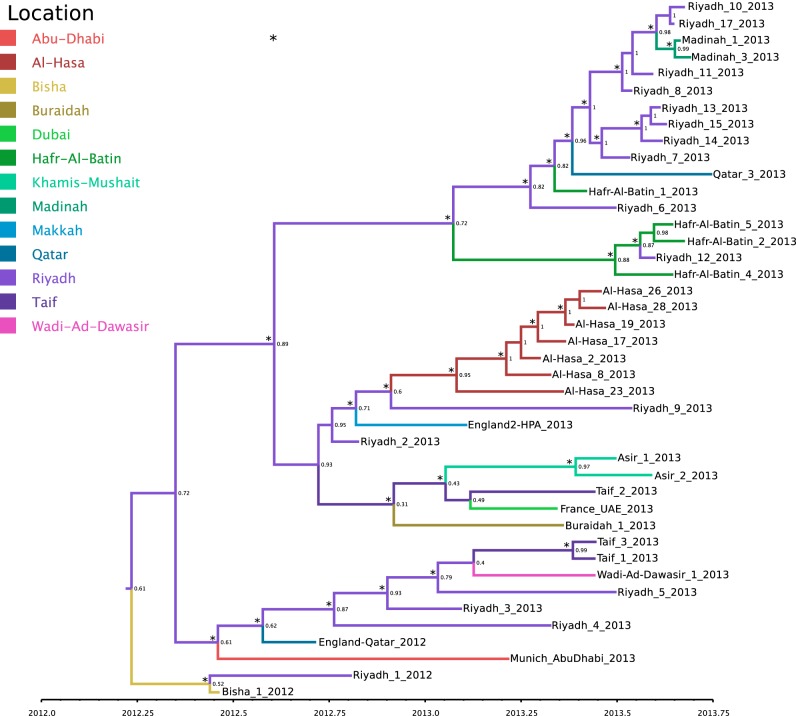
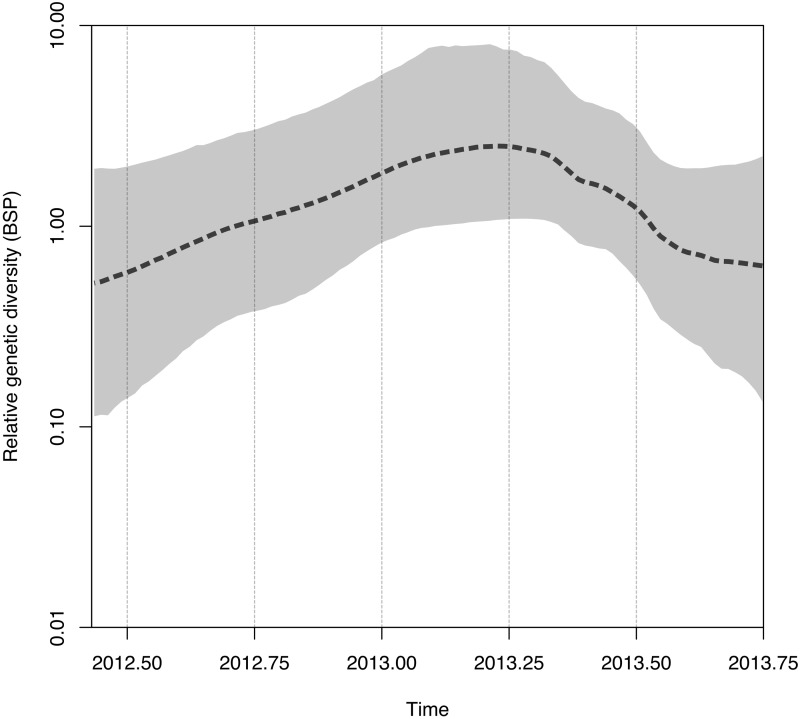
The Middle East respiratory syndrome coronavirus (MERS-CoV) was first documented in the Kingdom of Saudi Arabia (KSA) in 2012 and, to date, has been identified in 180 cases with 43% mortality. In this study, we have determined the MERS-CoV evolutionary rate, documented genetic variants of the virus and their distribution throughout the Arabian peninsula, and identified the genome positions under positive selection, important features for monitoring adaptation of MERS-CoV to human transmission and for identifying the source of infections. Respiratory samples from confirmed KSA MERS cases from May to September 2013 were subjected to whole-genome deep sequencing, and 32 complete or partial sequences (20 were ≥ 99% complete, 7 were 50 to 94% complete, and 5 were 27 to 50% complete) were obtained, bringing the total available MERS-CoV genomic sequences to 65. An evolutionary rate of 1.12 × 10(-3) substitutions per site per year (95% credible interval [95% CI], 8.76 × 10(-4); 1.37 × 10(-3)) was estimated, bringing the time to most recent common ancestor to March 2012 (95% CI, December 2011; June 2012). Only one MERS-CoV codon, spike 1020, located in a domain required for cell entry, is under strong positive selection. Four KSA MERS-CoV phylogenetic clades were found, with 3 clades apparently no longer contributing to current cases. The size of the population infected with MERS-CoV showed a gradual increase to June 2013, followed by a decline, possibly due to increased surveillance and infection control measures combined with a basic reproduction number (R0) for the virus that is less than 1.
Importance: MERS-CoV adaptation toward higher rates of sustained human-to-human transmission appears not to have occurred yet. While MERS-CoV transmission currently appears weak, careful monitoring of changes in MERS-CoV genomes and of the MERS epidemic should be maintained. The observation of phylogenetically related MERS-CoV in geographically diverse locations must be taken into account in efforts to identify the animal source and transmission of the virus.
Figures



Similar articles
-
Transmission and evolution of the Middle East respiratory syndrome coronavirus in Saudi Arabia: a descriptive genomic study.Lancet. 2013 Dec 14;382(9909):1993-2002. doi: 10.1016/S0140-6736(13)61887-5. Epub 2013 Sep 20. Lancet. 2013. PMID: 24055451 Free PMC article.
-
Rooting the phylogenetic tree of middle East respiratory syndrome coronavirus by characterization of a conspecific virus from an African bat.J Virol. 2014 Oct;88(19):11297-303. doi: 10.1128/JVI.01498-14. Epub 2014 Jul 16. J Virol. 2014. PMID: 25031349 Free PMC article.
-
Molecular Evolution and Structural Mapping of N-Terminal Domain in Spike Gene of Middle East Respiratory Syndrome Coronavirus (MERS-CoV).Viruses. 2020 May 2;12(5):502. doi: 10.3390/v12050502. Viruses. 2020. PMID: 32370153 Free PMC article.
-
MERS-CoV: epidemiology, molecular dynamics, therapeutics, and future challenges.Ann Clin Microbiol Antimicrob. 2021 Jan 18;20(1):8. doi: 10.1186/s12941-020-00414-7. Ann Clin Microbiol Antimicrob. 2021. PMID: 33461573 Free PMC article. Review.
-
Middle East respiratory syndrome coronavirus (MERS-CoV): evidence and speculations.Arch Virol. 2014 Jul;159(7):1575-84. doi: 10.1007/s00705-014-1995-5. Epub 2014 Feb 11. Arch Virol. 2014. PMID: 24515532 Free PMC article. Review.
Cited by
-
Middle East Respiratory Syndrome: Emergence of a Pathogenic Human Coronavirus.Annu Rev Med. 2017 Jan 14;68:387-399. doi: 10.1146/annurev-med-051215-031152. Epub 2016 Aug 26. Annu Rev Med. 2017. PMID: 27576010 Free PMC article. Review.
-
Identification of a Novel Betacoronavirus (Merbecovirus) in Amur Hedgehogs from China.Viruses. 2019 Oct 24;11(11):980. doi: 10.3390/v11110980. Viruses. 2019. PMID: 31653070 Free PMC article.
-
Prevalence and genetic diversity of coronaviruses in wild birds, Finland.Infect Ecol Epidemiol. 2017 Nov 28;7(1):1408360. doi: 10.1080/20008686.2017.1408360. eCollection 2017. Infect Ecol Epidemiol. 2017. PMID: 30788065 Free PMC article.
-
Coronaviruses - drug discovery and therapeutic options.Nat Rev Drug Discov. 2016 May;15(5):327-47. doi: 10.1038/nrd.2015.37. Epub 2016 Feb 12. Nat Rev Drug Discov. 2016. PMID: 26868298 Free PMC article. Review.
-
The Middle East Respiratory Syndrome Coronavirus - A Continuing Risk to Global Health Security.Adv Exp Med Biol. 2017;972:49-60. doi: 10.1007/5584_2016_133. Adv Exp Med Biol. 2017. PMID: 27966107 Free PMC article.
References
-
- van Boheemen S, de Graaf M, Lauber C, Bestebroer TM, Raj VS, Zaki AM, Osterhaus AD, Haagmans BL, Gorbalenya AE, Snijder EJ, Fouchier RA. 2012. Genomic characterization of a newly discovered coronavirus associated with acute respiratory distress syndrome in humans. mBio 3(6):e00473-12. 10.1128/mBio.00473-12 - DOI - PMC - PubMed
-
- Albarrak AM, Stephens GM, Hewson R, Memish ZA. 2012. Recovery from severe novel coronavirus infection. Saudi Med. J. 33:1265–1269 - PubMed
-
- WHO Accessed 27 January 2014. Middle East respiratory syndrome coronavirus (MERS-CoV). World Health Organization, Geneva, Switzerland: http://www.who.int/csr/don/2014_01_27mers/en/index.html
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
