

Re-entry into quiescence protects hematopoietic stem cells from the killing effect of chronic exposure to type I interferons
- PMID: 24493802
- PMCID: PMC3920566
- DOI: 10.1084/jem.20131043
Re-entry into quiescence protects hematopoietic stem cells from the killing effect of chronic exposure to type I interferons
Abstract
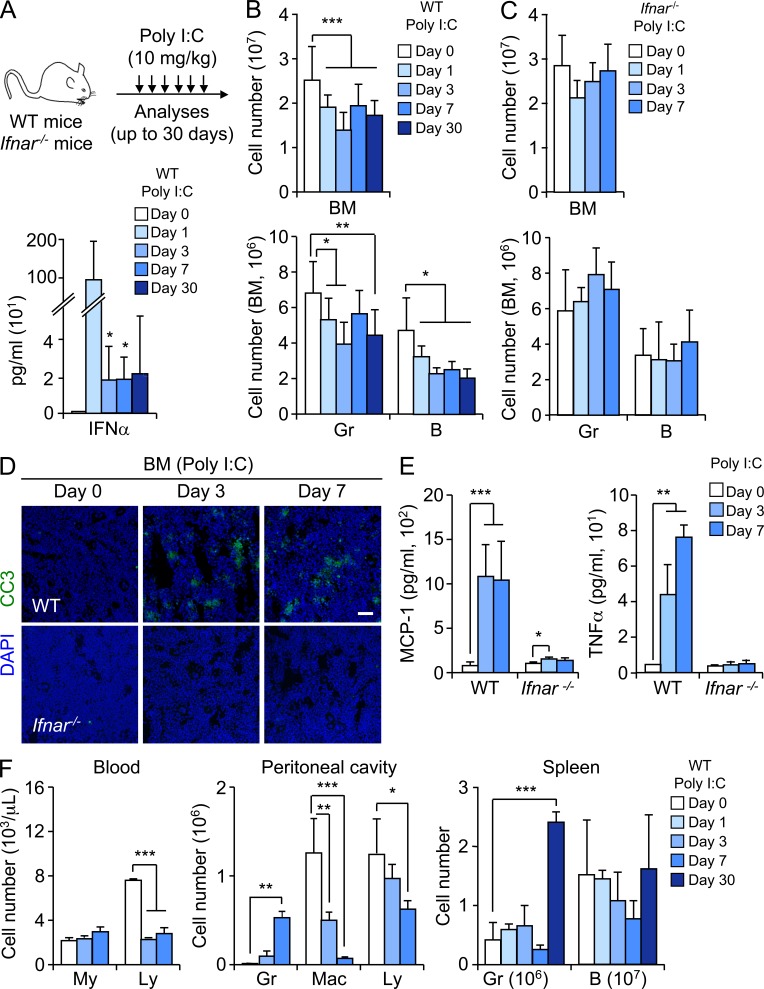
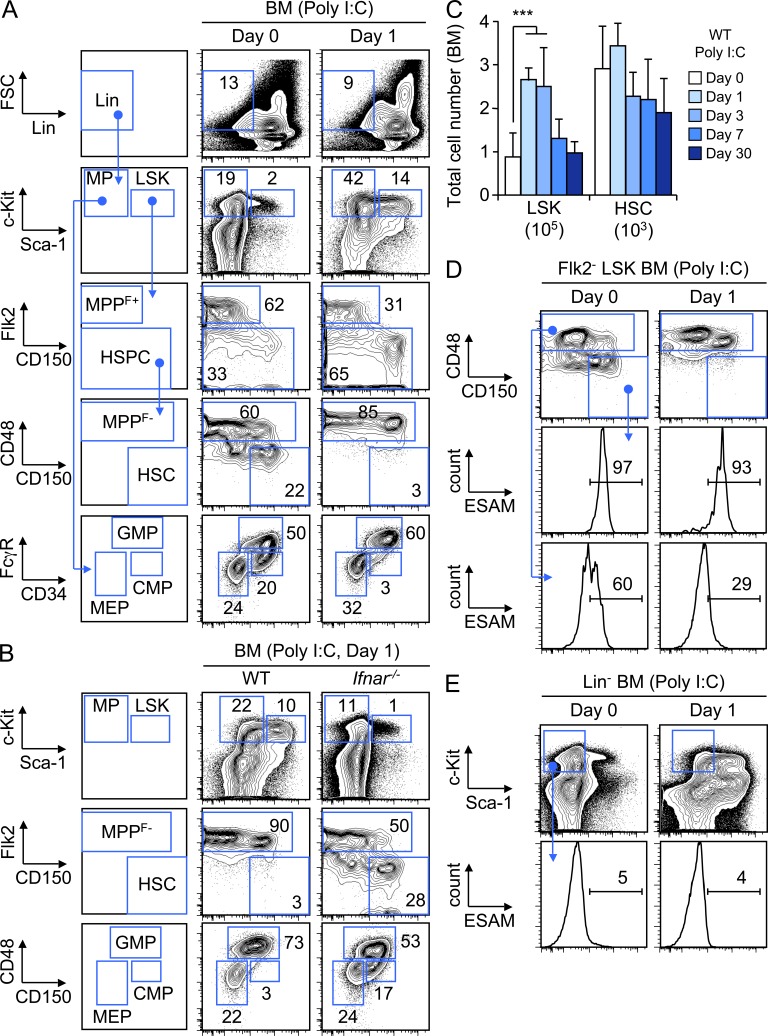
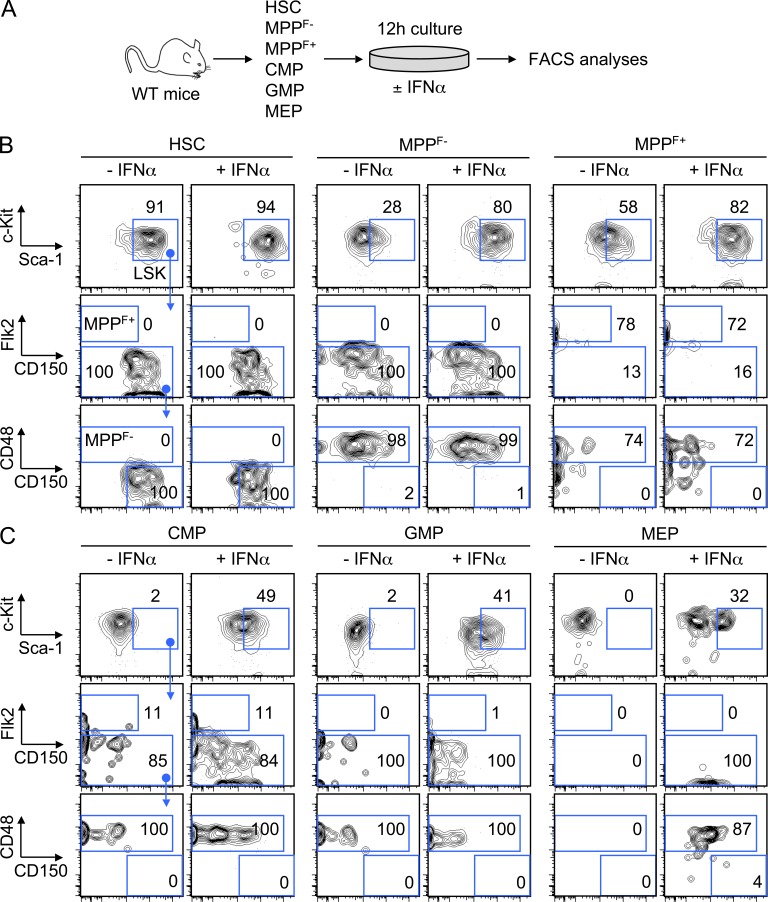
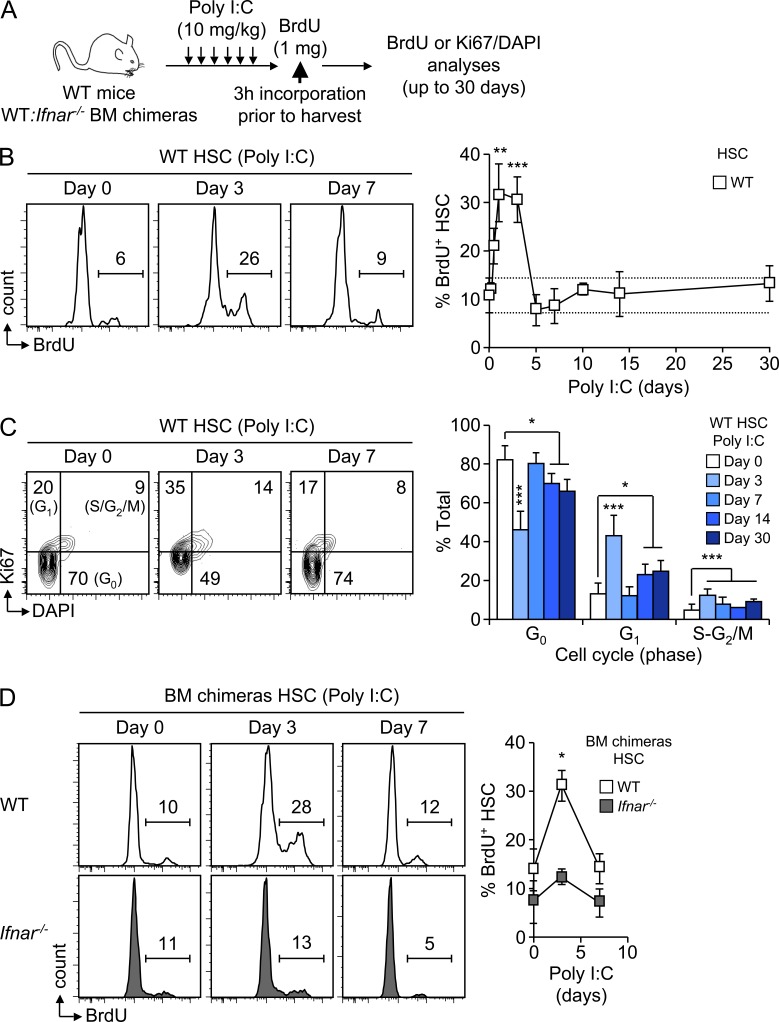
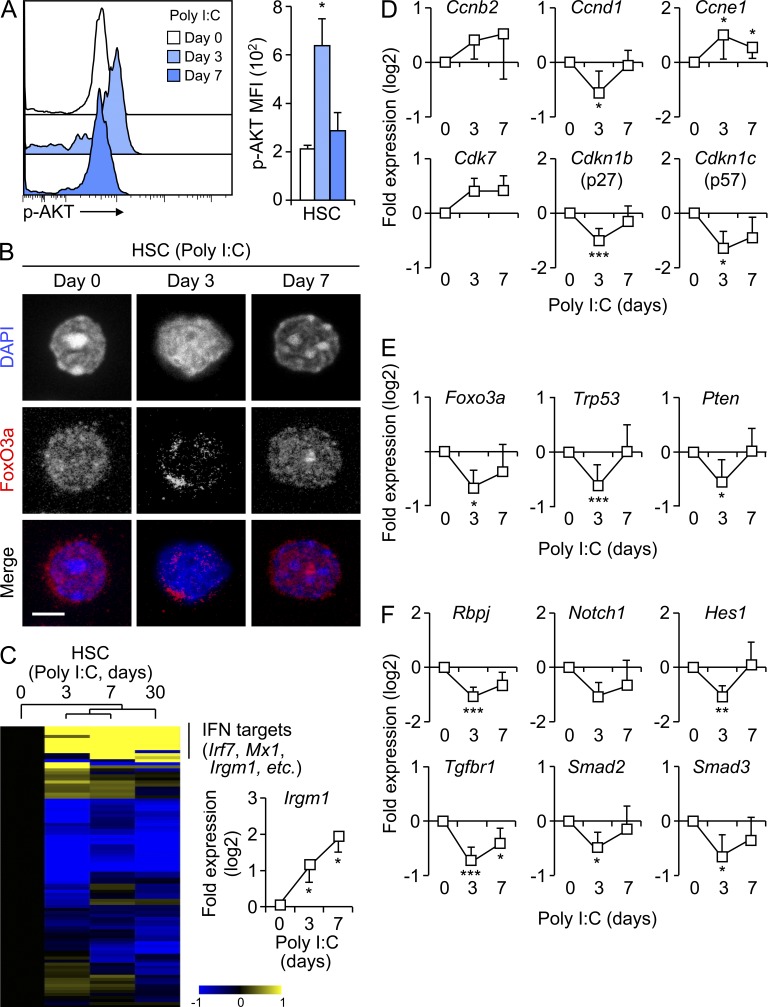
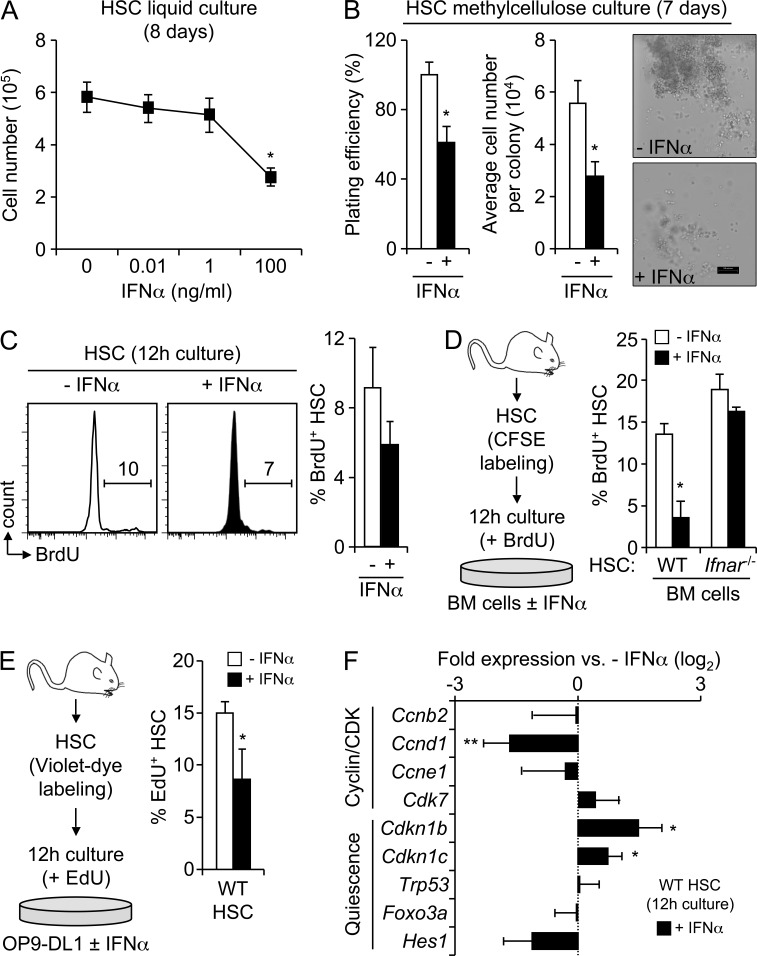
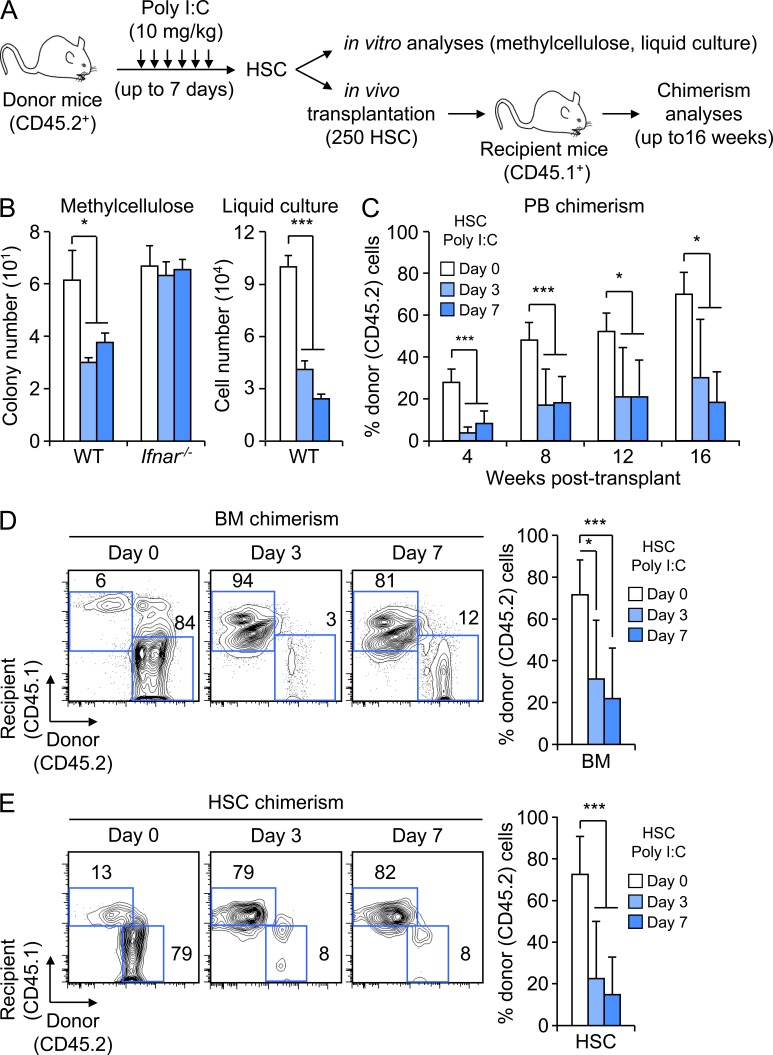
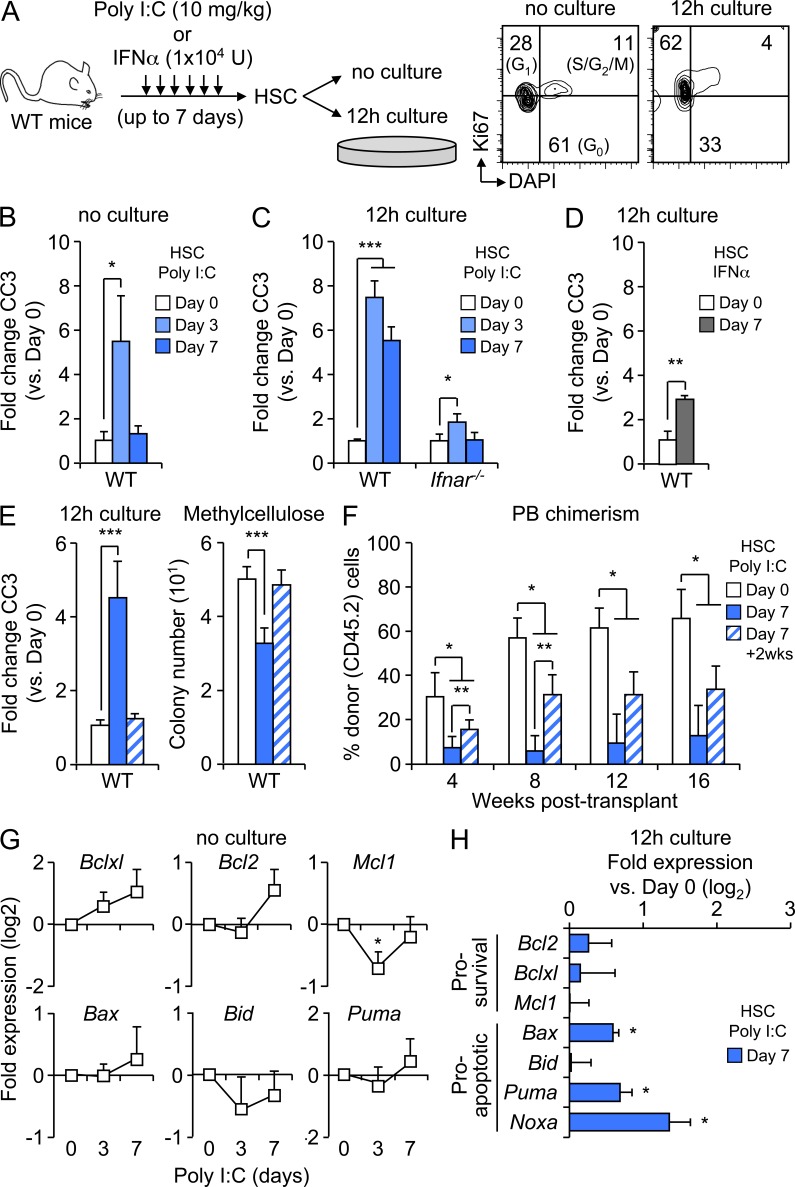
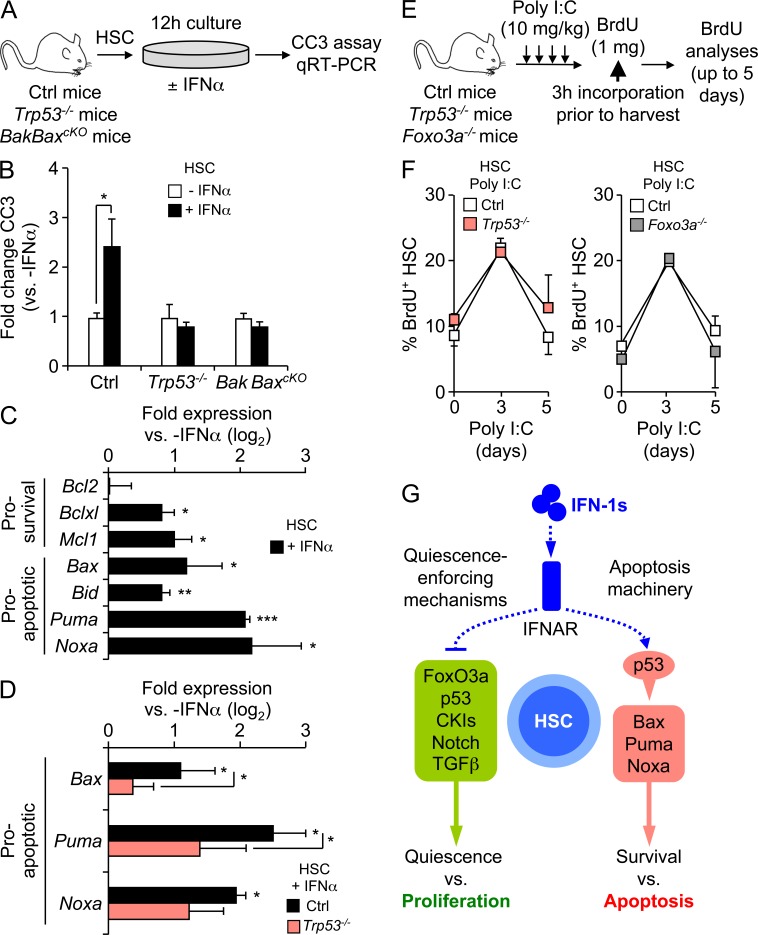
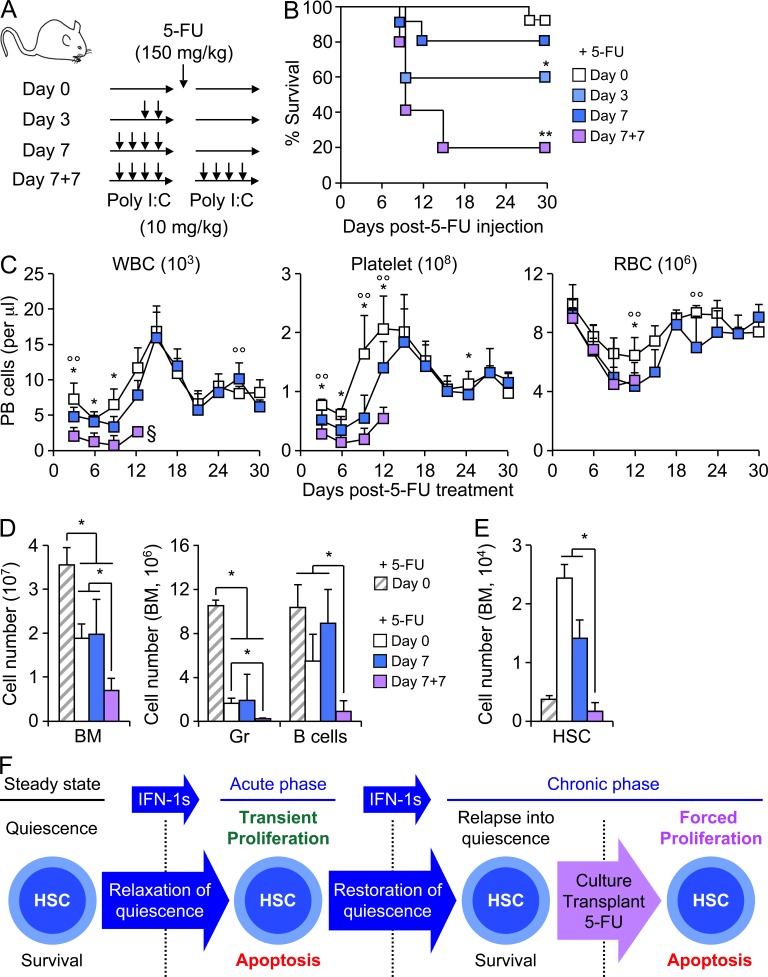
Type I interferons (IFN-1s) are antiviral cytokines that suppress blood production while paradoxically inducing hematopoietic stem cell (HSC) proliferation. Here, we clarify the relationship between the proliferative and suppressive effects of IFN-1s on HSC function during acute and chronic IFN-1 exposure. We show that IFN-1-driven HSC proliferation is a transient event resulting from a brief relaxation of quiescence-enforcing mechanisms in response to acute IFN-1 exposure, which occurs exclusively in vivo. We find that this proliferative burst fails to exhaust the HSC pool, which rapidly returns to quiescence in response to chronic IFN-1 exposure. Moreover, we demonstrate that IFN-1-exposed HSCs with reestablished quiescence are largely protected from the killing effects of IFNs unless forced back into the cell cycle due to culture, transplantation, or myeloablative treatment, at which point they activate a p53-dependent proapoptotic gene program. Collectively, our results demonstrate that quiescence acts as a safeguard mechanism to ensure survival of the HSC pool during chronic IFN-1 exposure. We show that IFN-1s can poise HSCs for apoptosis but induce direct cell killing only upon active proliferation, thereby establishing a mechanism for the suppressive effects of IFN-1s on HSC function.
Figures
Similar articles
-
Type I IFNs drive hematopoietic stem and progenitor cell collapse via impaired proliferation and increased RIPK1-dependent cell death during shock-like ehrlichial infection.PLoS Pathog. 2018 Aug 6;14(8):e1007234. doi: 10.1371/journal.ppat.1007234. eCollection 2018 Aug. PLoS Pathog. 2018. PMID: 30080899 Free PMC article.
-
Interferon regulatory factor-2 protects quiescent hematopoietic stem cells from type I interferon-dependent exhaustion.Nat Med. 2009 Jun;15(6):696-700. doi: 10.1038/nm.1973. Nat Med. 2009. PMID: 19483695
-
Adiponectin Enhances Quiescence Exit of Murine Hematopoietic Stem Cells and Hematopoietic Recovery Through mTORC1 Potentiation.Stem Cells. 2017 Jul;35(7):1835-1848. doi: 10.1002/stem.2640. Epub 2017 May 23. Stem Cells. 2017. PMID: 28480607
-
Newly emerging roles for prostaglandin E2 regulation of hematopoiesis and hematopoietic stem cell engraftment.Curr Opin Hematol. 2010 Jul;17(4):308-12. doi: 10.1097/MOH.0b013e32833a888c. Curr Opin Hematol. 2010. PMID: 20473159 Review.
-
The p53 tumor suppressor protein is a critical regulator of hematopoietic stem cell behavior.Cell Cycle. 2009 Oct 1;8(19):3120-4. doi: 10.4161/cc.8.19.9627. Cell Cycle. 2009. PMID: 19755852 Free PMC article. Review.
Cited by
-
Hematopoietic stem cell heterogeneity is linked to the initiation and therapeutic response of myeloproliferative neoplasms.Cell Stem Cell. 2021 Apr 1;28(4):780. doi: 10.1016/j.stem.2021.02.026. Cell Stem Cell. 2021. PMID: 33798424 Free PMC article.
-
Innate Immune Memory in Hematopoietic Stem/Progenitor Cells: Myeloid-Biased Differentiation and the Role of Interferon.Front Immunol. 2021 Mar 29;12:621333. doi: 10.3389/fimmu.2021.621333. eCollection 2021. Front Immunol. 2021. PMID: 33854500 Free PMC article. Review.
-
Hematopoietic responses to SARS-CoV-2 infection.Cell Mol Life Sci. 2022 Mar 13;79(3):187. doi: 10.1007/s00018-022-04220-6. Cell Mol Life Sci. 2022. PMID: 35284964 Free PMC article. Review.
-
Distinct effects of ruxolitinib and interferon-alpha on murine JAK2V617F myeloproliferative neoplasm hematopoietic stem cell populations.Leukemia. 2020 Apr;34(4):1075-1089. doi: 10.1038/s41375-019-0638-y. Epub 2019 Nov 15. Leukemia. 2020. PMID: 31732720 Free PMC article.
-
Type 1 IFN and PD-L1 Coordinate Lymphatic Endothelial Cell Expansion and Contraction during an Inflammatory Immune Response.J Immunol. 2018 Sep 15;201(6):1735-1747. doi: 10.4049/jimmunol.1800271. Epub 2018 Jul 25. J Immunol. 2018. PMID: 30045970 Free PMC article.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
