

DAXX co-folds with H3.3/H4 using high local stability conferred by the H3.3 variant recognition residues
- PMID: 24493739
- PMCID: PMC3985662
- DOI: 10.1093/nar/gku090
DAXX co-folds with H3.3/H4 using high local stability conferred by the H3.3 variant recognition residues
Abstract
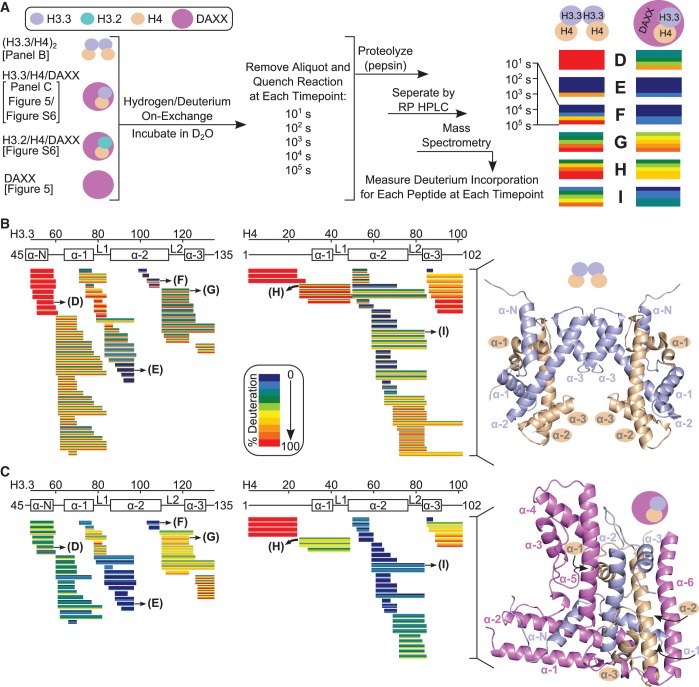
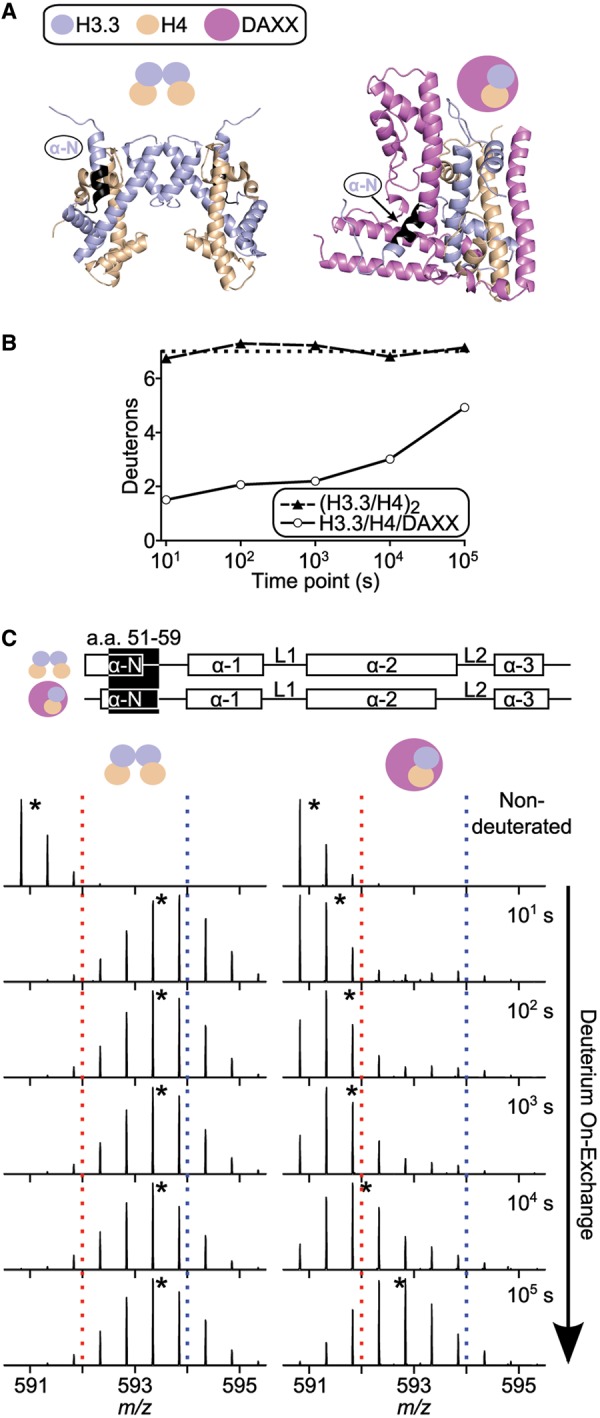
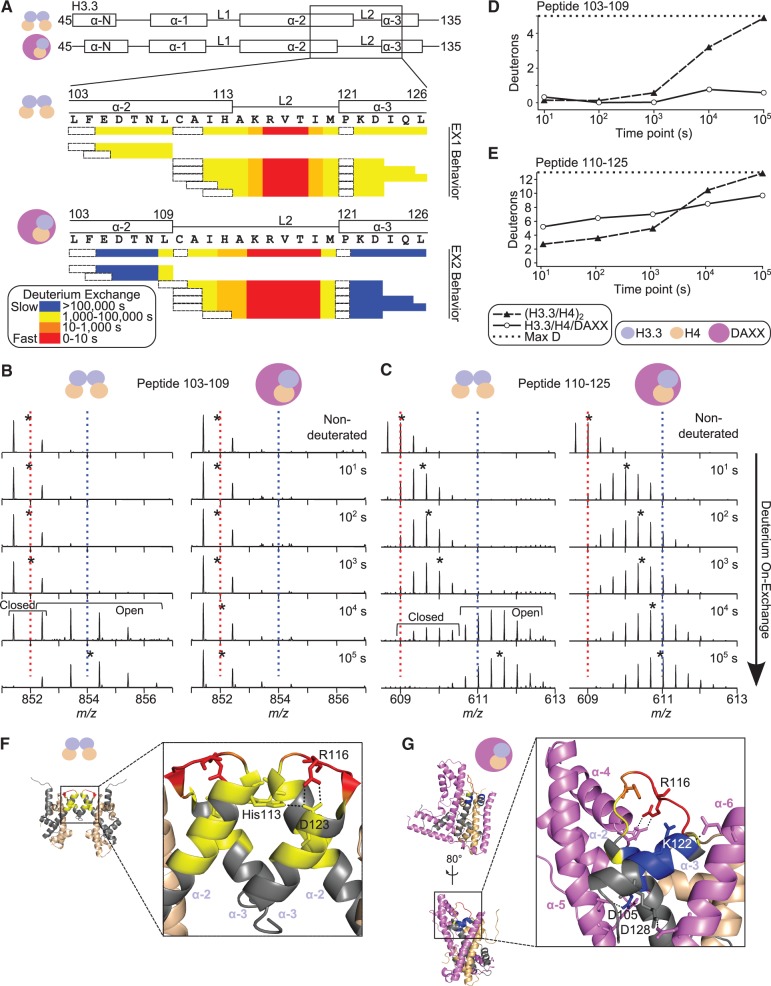
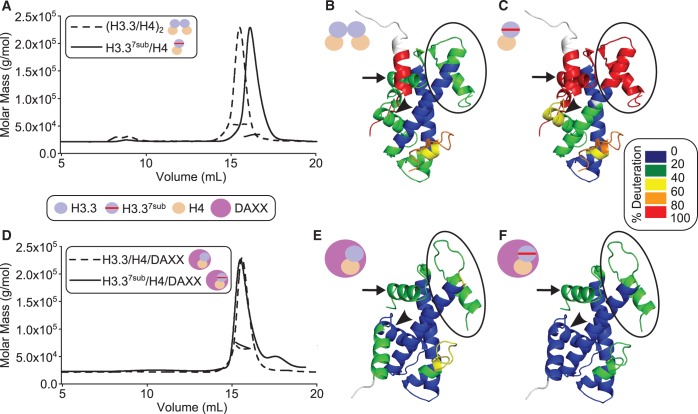
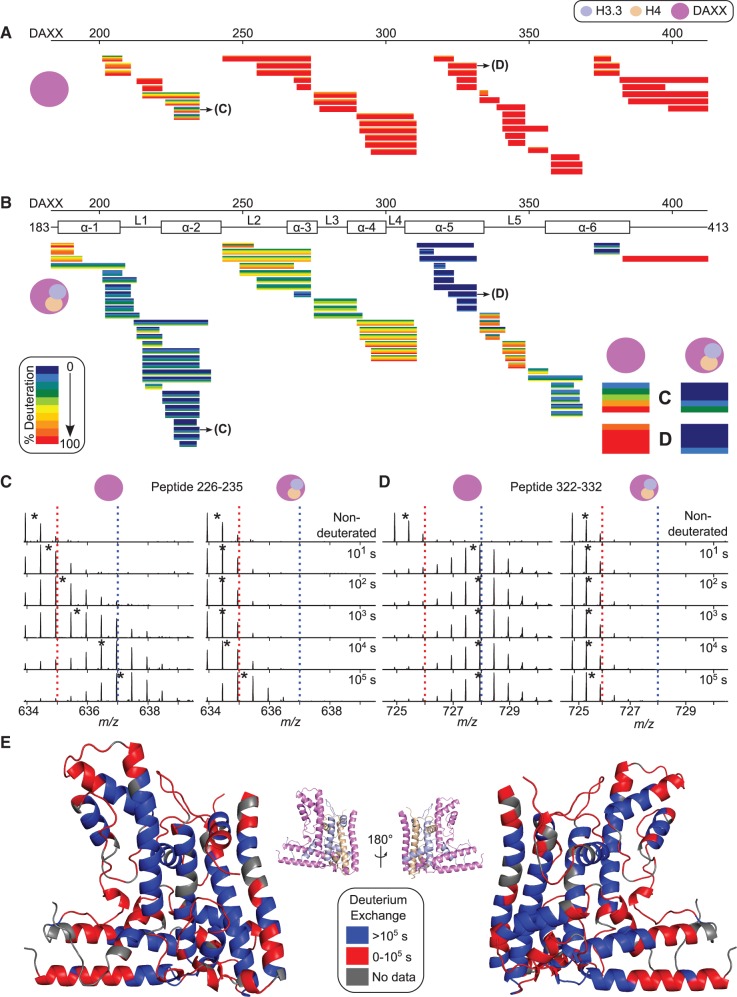
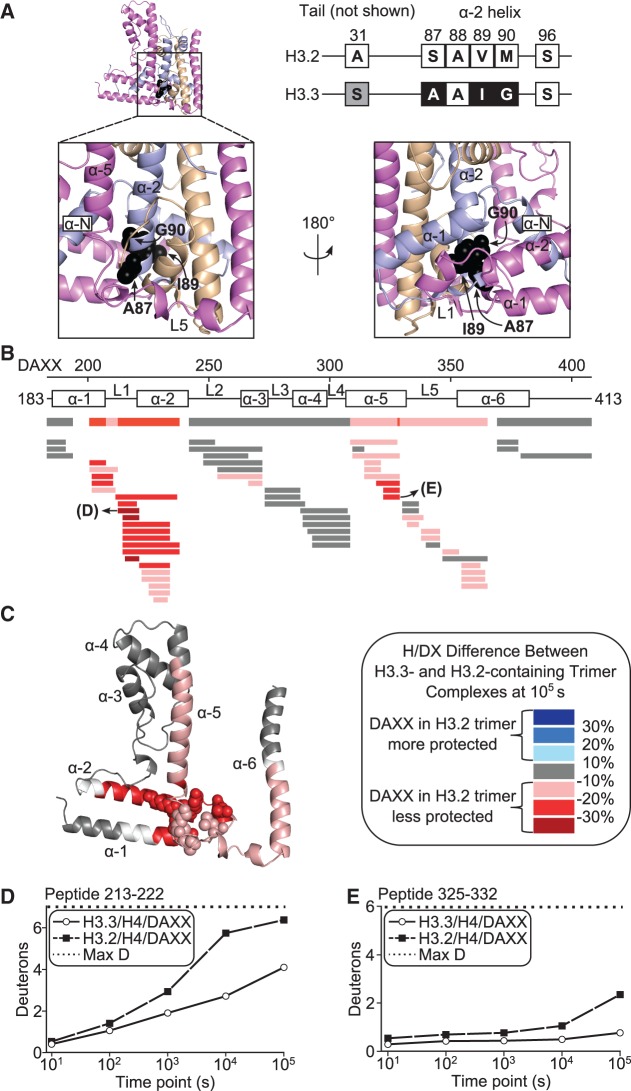
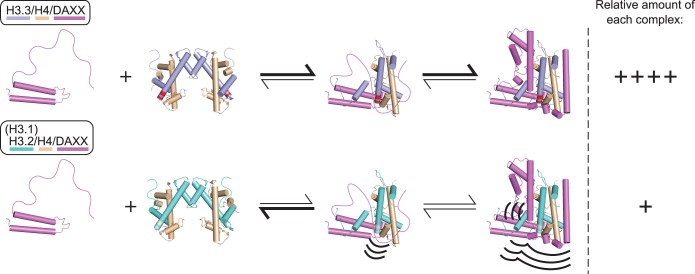
Histone chaperones are a diverse class of proteins that facilitate chromatin assembly. Their ability to stabilize highly abundant histone proteins in the cellular environment prevents non-specific interactions and promotes nucleosome formation, but the various mechanisms for doing so are not well understood. We now focus on the dynamic features of the DAXX histone chaperone that have been elusive from previous structural studies. Using hydrogen/deuterium exchange coupled to mass spectrometry (H/DX-MS), we elucidate the concerted binding-folding of DAXX with histone variants H3.3/H4 and H3.2/H4 and find that high local stability at the variant-specific recognition residues rationalizes its known selectivity for H3.3. We show that the DAXX histone binding domain is largely disordered in solution and that formation of the H3.3/H4/DAXX complex induces folding and dramatic global stabilization of both histone and chaperone. Thus, DAXX uses a novel strategy as a molecular chaperone that paradoxically couples its own folding to substrate recognition and binding. Further, we propose a model for the chromatin assembly reaction it mediates, including a stepwise folding pathway that helps explain the fidelity of DAXX in associating with the H3.3 variant, despite an extensive and nearly identical binding surface on its counterparts, H3.1 and H3.2.
Figures
Similar articles
-
DAXX envelops a histone H3.3-H4 dimer for H3.3-specific recognition.Nature. 2012 Nov 22;491(7425):560-5. doi: 10.1038/nature11608. Epub 2012 Oct 17. Nature. 2012. PMID: 23075851 Free PMC article.
-
Energy landscape quantifications of histone H3.3 recognition by chaperone DAXX reveal an uncoupled binding specificity and affinity.Phys Chem Chem Phys. 2023 Oct 25;25(41):27981-27993. doi: 10.1039/d3cp02612d. Phys Chem Chem Phys. 2023. PMID: 37818851
-
H3.Y discriminates between HIRA and DAXX chaperone complexes and reveals unexpected insights into human DAXX-H3.3-H4 binding and deposition requirements.Nucleic Acids Res. 2017 Jun 2;45(10):5691-5706. doi: 10.1093/nar/gkx131. Nucleic Acids Res. 2017. PMID: 28334823 Free PMC article.
-
H3-H4 Histone Chaperone Pathways.Annu Rev Genet. 2018 Nov 23;52:109-130. doi: 10.1146/annurev-genet-120417-031547. Epub 2018 Sep 5. Annu Rev Genet. 2018. PMID: 30183406 Review.
-
A Molecular Prospective for HIRA Complex Assembly and H3.3-Specific Histone Chaperone Function.J Mol Biol. 2017 Jun 30;429(13):1924-1933. doi: 10.1016/j.jmb.2016.11.010. Epub 2016 Nov 19. J Mol Biol. 2017. PMID: 27871933 Free PMC article. Review.
Cited by
-
The roles of histone variants in fine-tuning chromatin organization and function.Nat Rev Mol Cell Biol. 2020 Sep;21(9):522-541. doi: 10.1038/s41580-020-0262-8. Epub 2020 Jul 14. Nat Rev Mol Cell Biol. 2020. PMID: 32665685 Free PMC article. Review.
-
Histone chaperone networks shaping chromatin function.Nat Rev Mol Cell Biol. 2017 Mar;18(3):141-158. doi: 10.1038/nrm.2016.159. Epub 2017 Jan 5. Nat Rev Mol Cell Biol. 2017. PMID: 28053344 Free PMC article. Review.
-
Maintaining memory of silencing at imprinted differentially methylated regions.Cell Mol Life Sci. 2016 May;73(9):1871-9. doi: 10.1007/s00018-016-2157-6. Epub 2016 Feb 16. Cell Mol Life Sci. 2016. PMID: 26883803 Free PMC article. Review.
-
DNA damage-induced regulatory interplay between DAXX, p53, ATM kinase and Wip1 phosphatase.Cell Cycle. 2015;14(3):375-87. doi: 10.4161/15384101.2014.988019. Cell Cycle. 2015. PMID: 25659035 Free PMC article.
-
Chasing Tails: Cathepsin-L Improves Structural Analysis of Histones by HX-MS.Mol Cell Proteomics. 2019 Oct;18(10):2089-2098. doi: 10.1074/mcp.RA119.001325. Epub 2019 Aug 13. Mol Cell Proteomics. 2019. PMID: 31409669 Free PMC article.
References
-
- Hondele M, Ladurner AG. The chaperone-histone partnership: for the greater good of histone traffic and chromatin plasticity. Curr. Opin. Struct. Biol. 2011;21:698–708. - PubMed
-
- Ray-Gallet D, Quivy J-P, Scamps C, Martini EM-D, Lipinski M, Almouzni G. HIRA is critical for a nucleosome assembly pathway independent of DNA synthesis. Mol. Cell. 2002;9:1091–1100. - PubMed
-
- Tagami H, Ray-Gallet D, Almouzni G, Nakatani Y. Histone H3.1 and H3.3 complexes mediate nucleosome assembly pathways dependent or independent of DNA synthesis. Cell. 2004;116:51–61. - PubMed
-
- Dunleavy EM, Roche D, Tagami H, Lacoste N, Ray-Gallet D, Nakamura Y, Daigo Y, Nakatani Y, Almouzni-Pettinotti G. HJURP is a cell-cycle-dependent maintenance and deposition factor of CENP-A at centromeres. Cell. 2009;137:485–497. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
