

Structural insights into competitive antagonism in NMDA receptors
- PMID: 24462099
- PMCID: PMC3997178
- DOI: 10.1016/j.neuron.2013.11.033
Structural insights into competitive antagonism in NMDA receptors
Abstract
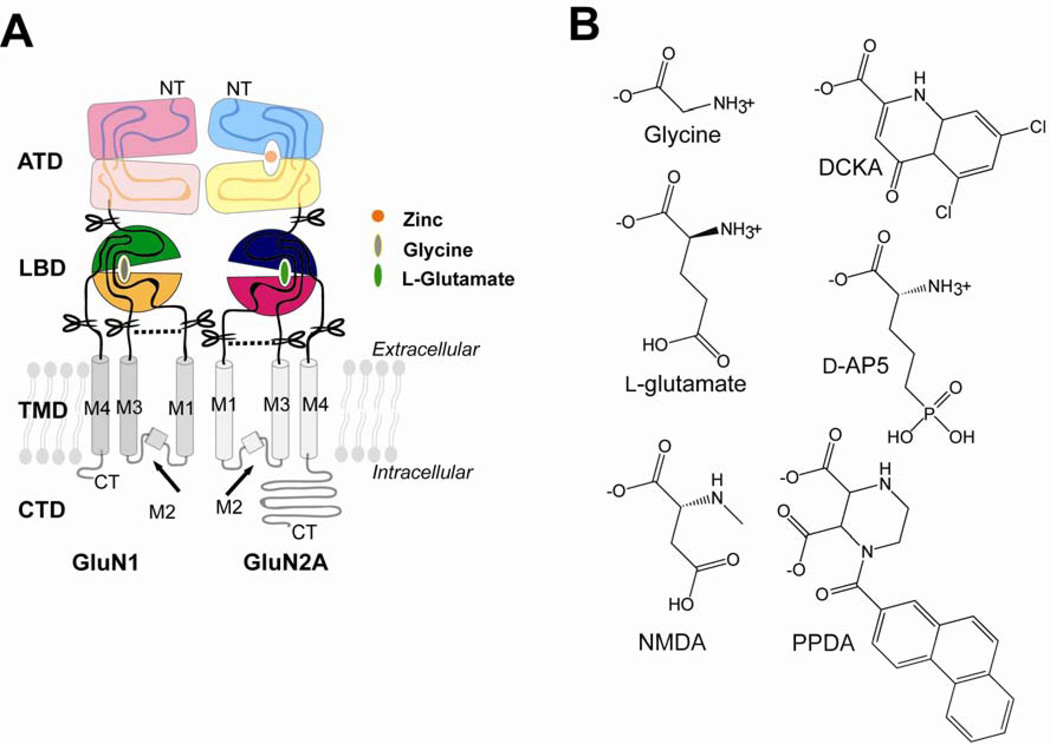
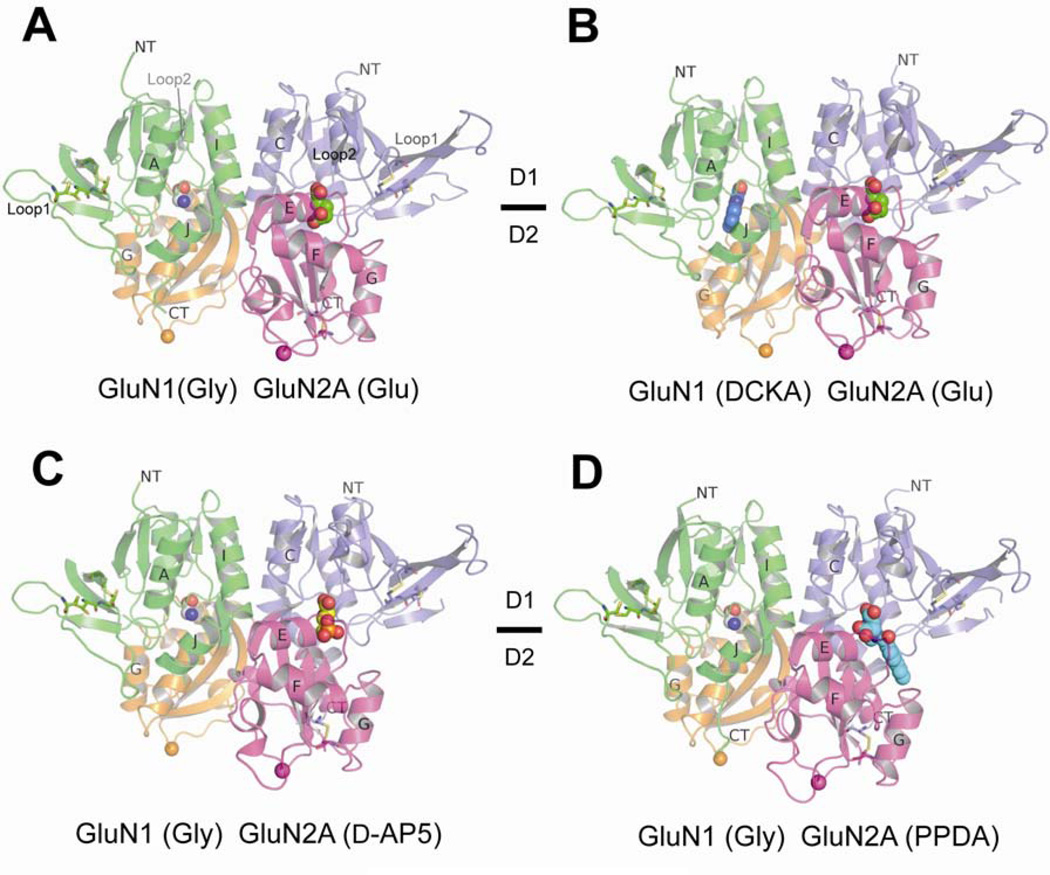
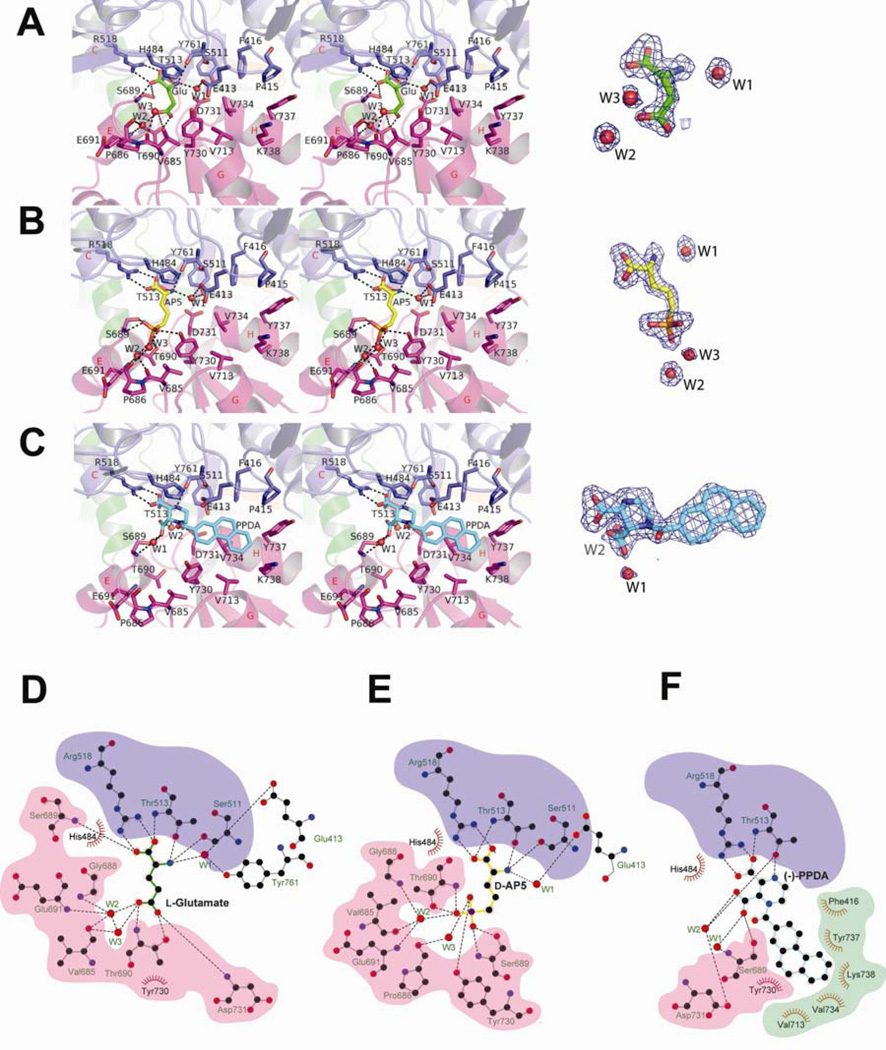
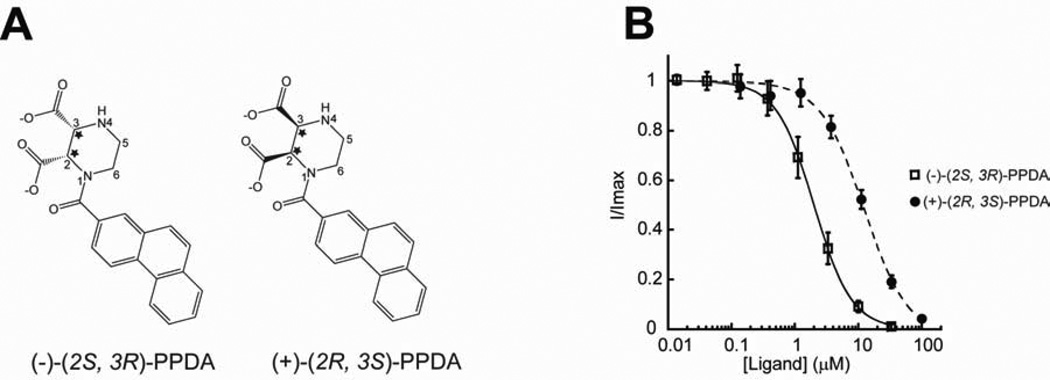
There has been a great level of enthusiasm to downregulate overactive N-methyl-D-aspartate (NMDA) receptors to protect neurons from excitotoxicity. NMDA receptors play pivotal roles in basic brain development and functions as well as in neurological disorders and diseases. However, mechanistic understanding of antagonism in NMDA receptors is limited due to complete lack of antagonist-bound structures for the L-glutamate-binding GluN2 subunits. Here, we report the crystal structures of GluN1/GluN2A NMDA receptor ligand-binding domain (LBD) heterodimers in complex with GluN1- and GluN2-targeting antagonists. The crystal structures reveal that the antagonists, D-(-)-2-amino-5-phosphonopentanoic acid (D-AP5) and 1-(phenanthrene-2-carbonyl)piperazine-2,3-dicarboxylic acid (PPDA), have discrete binding modes and mechanisms for opening of the bilobed architecture of GluN2A LBD compared to the agonist-bound form. The current study shows distinct ways by which the conformations of NMDA receptor LBDs may be controlled and coupled to receptor inhibition and provides possible strategies to develop therapeutic compounds with higher subtype-specificity.
Copyright © 2014 Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Structure-based discovery of antagonists for GluN3-containing N-methyl-D-aspartate receptors.Neuropharmacology. 2013 Dec;75:324-36. doi: 10.1016/j.neuropharm.2013.08.003. Epub 2013 Aug 22. Neuropharmacology. 2013. PMID: 23973313 Free PMC article.
-
Structural basis of subunit selectivity for competitive NMDA receptor antagonists with preference for GluN2A over GluN2B subunits.Proc Natl Acad Sci U S A. 2017 Aug 15;114(33):E6942-E6951. doi: 10.1073/pnas.1707752114. Epub 2017 Jul 31. Proc Natl Acad Sci U S A. 2017. PMID: 28760974 Free PMC article.
-
Arrangement of subunits in functional NMDA receptors.J Neurosci. 2011 Aug 3;31(31):11295-304. doi: 10.1523/JNEUROSCI.5612-10.2011. J Neurosci. 2011. PMID: 21813689 Free PMC article.
-
(1S,2R)-1-Phenyl-2-[(S)-1-aminopropyl]-N,N-diethylcyclopropanecarboxamide (PPDC), a new class of NMDA-receptor antagonist: molecular design by a novel conformational restriction strategy.Jpn J Pharmacol. 2001 Mar;85(3):207-13. doi: 10.1254/jjp.85.207. Jpn J Pharmacol. 2001. PMID: 11325012 Review.
-
Discovery of GluN2A subtype-selective N-methyl-d-aspartate (NMDA) receptor ligands.Acta Pharm Sin B. 2024 May;14(5):1987-2005. doi: 10.1016/j.apsb.2024.01.004. Epub 2024 Jan 10. Acta Pharm Sin B. 2024. PMID: 38799621 Free PMC article. Review.
Cited by
-
Activation of NMDA receptors and the mechanism of inhibition by ifenprodil.Nature. 2016 Jun 2;534(7605):63-8. doi: 10.1038/nature17679. Epub 2016 May 2. Nature. 2016. PMID: 27135925 Free PMC article.
-
The GluN2B-Glu413Gly NMDA receptor variant arising from a de novo GRIN2B mutation promotes ligand-unbinding and domain opening.Proteins. 2018 Dec;86(12):1265-1276. doi: 10.1002/prot.25595. Epub 2018 Oct 5. Proteins. 2018. PMID: 30168177 Free PMC article.
-
Deeper Insights into the Allosteric Modulation of Ionotropic Glutamate Receptors.Neuron. 2016 Sep 21;91(6):1187-1189. doi: 10.1016/j.neuron.2016.09.013. Neuron. 2016. PMID: 27657445 Free PMC article.
-
Prediction of N-Methyl-D-Aspartate Receptor GluN1-Ligand Binding Affinity by a Novel SVM-Pose/SVM-Score Combinatorial Ensemble Docking Scheme.Sci Rep. 2017 Jan 6;7:40053. doi: 10.1038/srep40053. Sci Rep. 2017. PMID: 28059133 Free PMC article.
-
Structure and function of GluN1-3A NMDA receptor excitatory glycine receptor channel.Sci Adv. 2024 Apr 12;10(15):eadl5952. doi: 10.1126/sciadv.adl5952. Epub 2024 Apr 10. Sci Adv. 2024. PMID: 38598639 Free PMC article.
References
-
- Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 2002;58:1948–1954. - PubMed
-
- Armstrong N, Gouaux E. Mechanisms for activation and antagonism of an AMPA-sensitive glutamate receptor: crystal structures of the GluR2 ligand binding core. Neuron. 2000;28:165–181. - PubMed
-
- Bettini E, Sava A, Griffante C, Carignani C, Buson A, Capelli AM, Negri M, Andreetta F, Senar-Sancho SA, Guiral L, et al. Identification and characterization of novel NMDA receptor antagonists selective for NR2A- over NR2B-containing receptors. J Pharmacol Exp Ther. 2010;335:636–644. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
