

Inhibition of endothelial FAK activity prevents tumor metastasis by enhancing barrier function
- PMID: 24446483
- PMCID: PMC3897185
- DOI: 10.1083/jcb.201307067
Inhibition of endothelial FAK activity prevents tumor metastasis by enhancing barrier function
Abstract
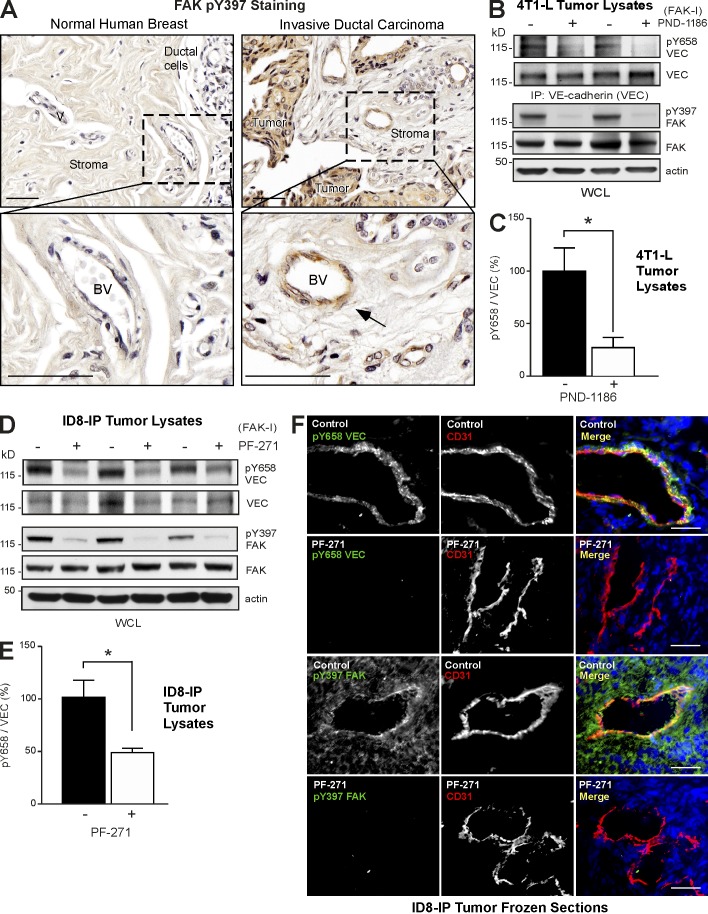
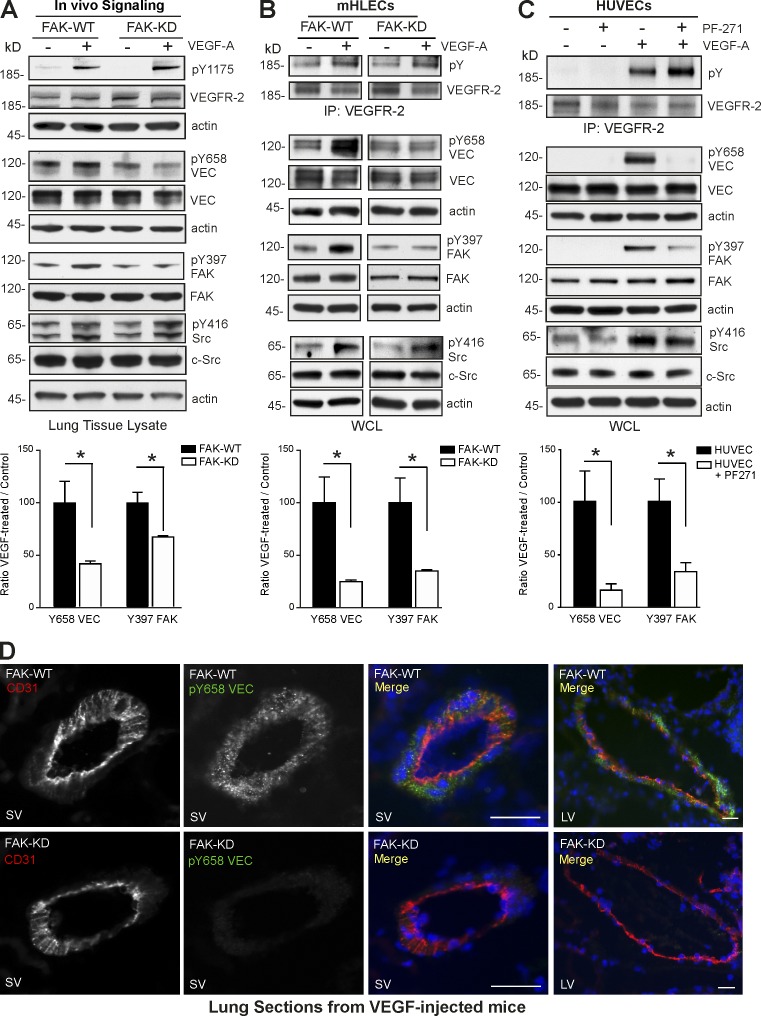
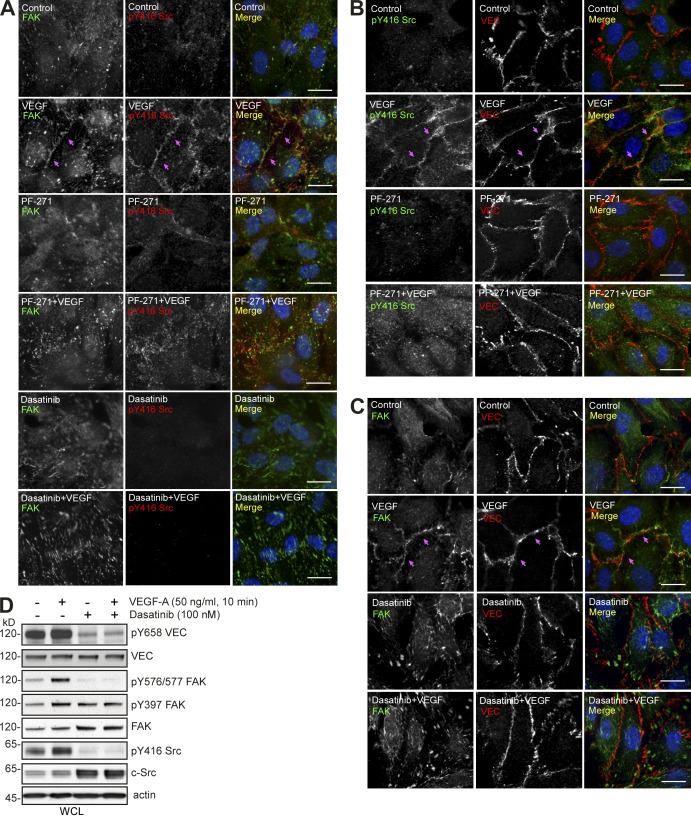
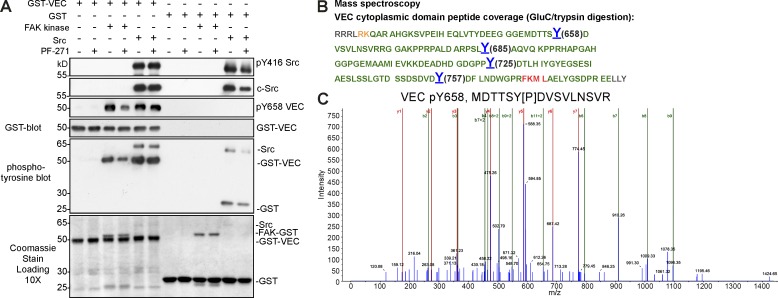
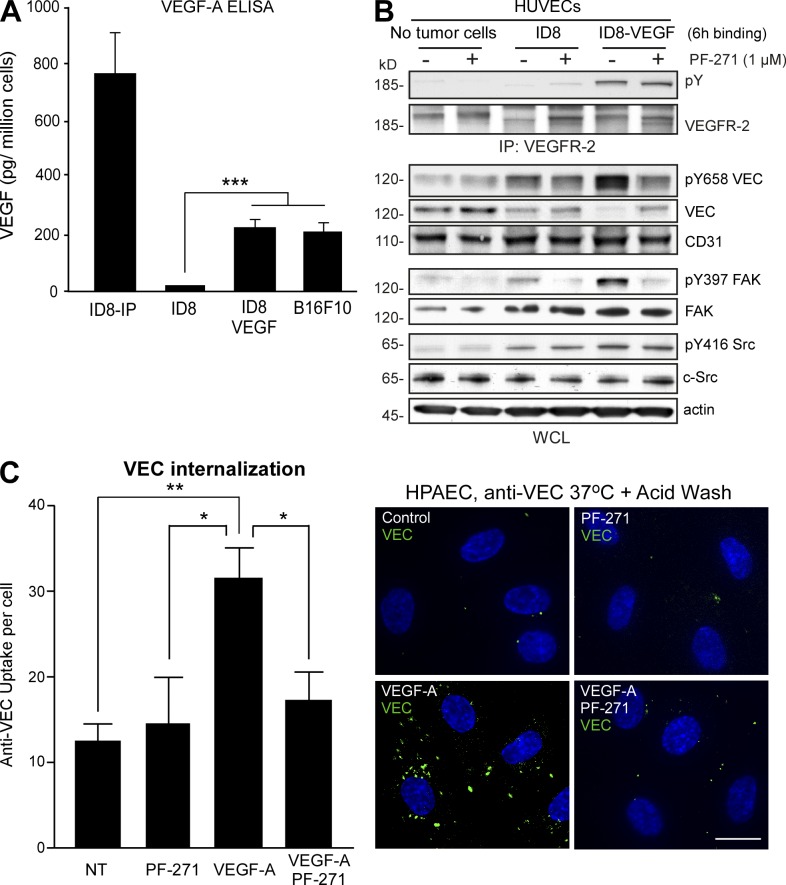
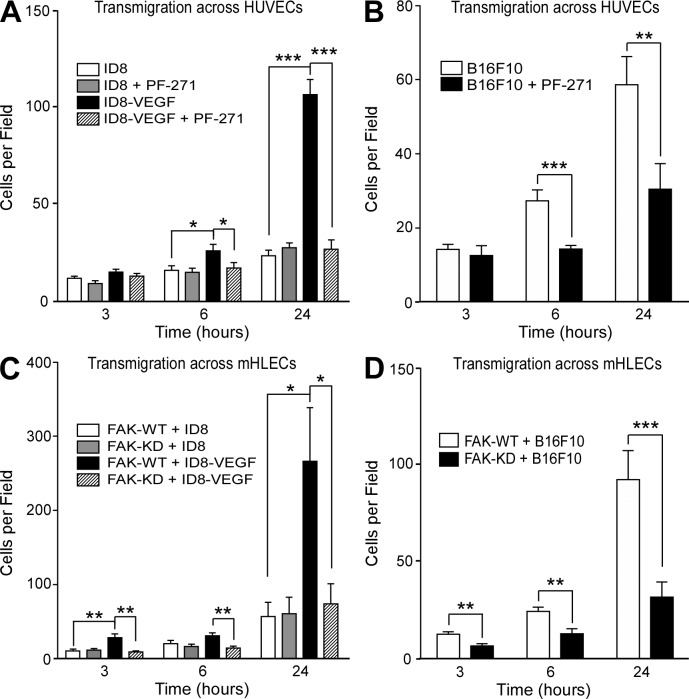
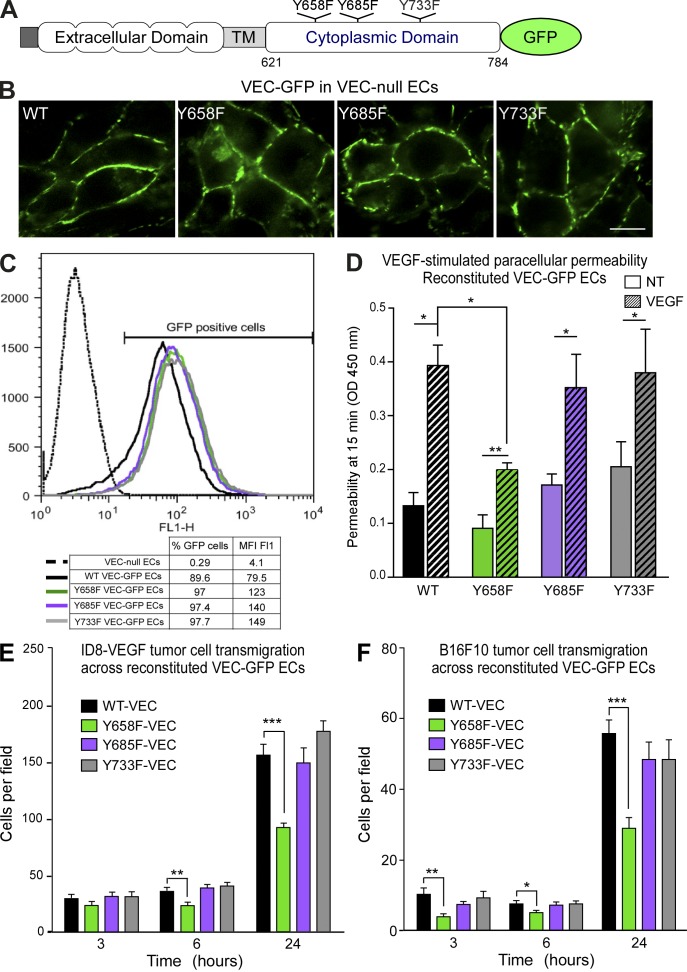
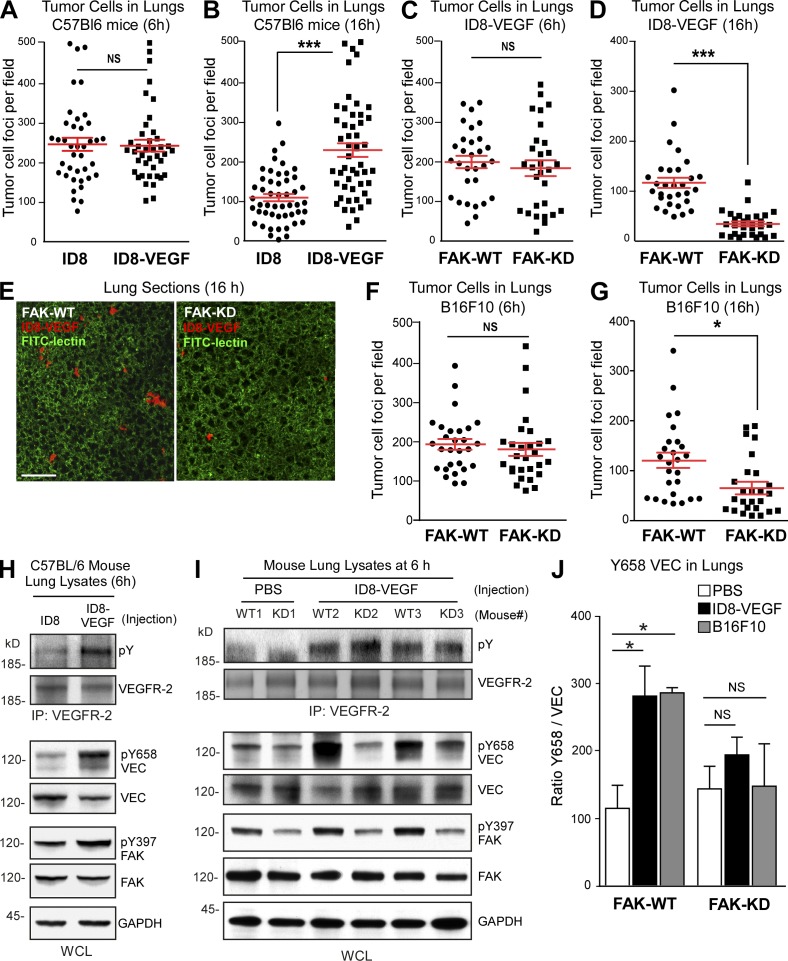
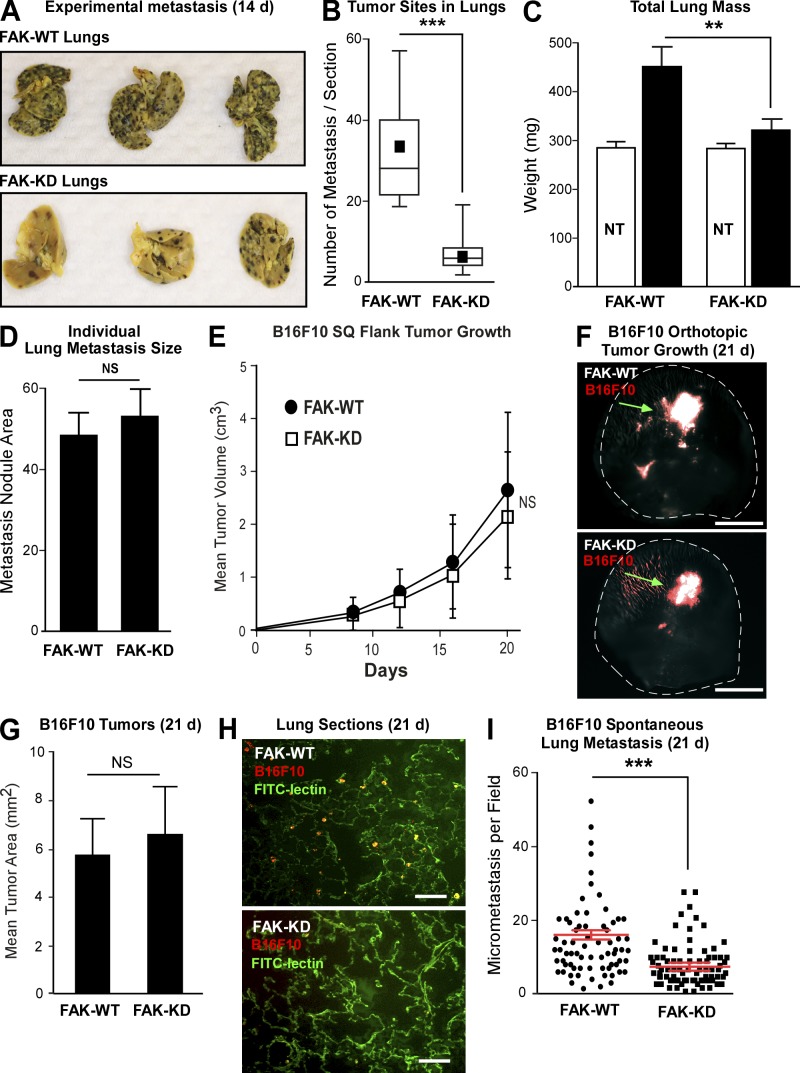
Pharmacological focal adhesion kinase (FAK) inhibition prevents tumor growth and metastasis, via actions on both tumor and stromal cells. In this paper, we show that vascular endothelial cadherin (VEC) tyrosine (Y) 658 is a target of FAK in tumor-associated endothelial cells (ECs). Conditional kinase-dead FAK knockin within ECs inhibited recombinant vascular endothelial growth factor (VEGF-A) and tumor-induced VEC-Y658 phosphorylation in vivo. Adherence of VEGF-expressing tumor cells to ECs triggered FAK-dependent VEC-Y658 phosphorylation. Both FAK inhibition and VEC-Y658F mutation within ECs prevented VEGF-initiated paracellular permeability and tumor cell transmigration across EC barriers. In mice, EC FAK inhibition prevented VEGF-dependent tumor cell extravasation and melanoma dermal to lung metastasis without affecting primary tumor growth. As pharmacological c-Src or FAK inhibition prevents VEGF-stimulated c-Src and FAK translocation to EC adherens junctions, but FAK inhibition does not alter c-Src activation, our experiments identify EC FAK as a key intermediate between c-Src and the regulation of EC barrier function controlling tumor metastasis.
Figures
Similar articles
-
VEGF-induced vascular permeability is mediated by FAK.Dev Cell. 2012 Jan 17;22(1):146-57. doi: 10.1016/j.devcel.2011.11.002. Dev Cell. 2012. PMID: 22264731 Free PMC article.
-
Matrix stiffness regulates vascular integrity through focal adhesion kinase activity.FASEB J. 2019 Jan;33(1):1199-1208. doi: 10.1096/fj.201800841R. Epub 2018 Aug 13. FASEB J. 2019. PMID: 30102569 Free PMC article.
-
Temporal Dynamics of VEGFA-Induced VEGFR2/FAK Co-Localization Depend on SHB.Cells. 2019 Dec 15;8(12):1645. doi: 10.3390/cells8121645. Cells. 2019. PMID: 31847469 Free PMC article.
-
Endothelial barrier disruption by VEGF-mediated Src activity potentiates tumor cell extravasation and metastasis.J Cell Biol. 2004 Oct 25;167(2):223-9. doi: 10.1083/jcb.200408130. J Cell Biol. 2004. PMID: 15504909 Free PMC article.
-
Molecular Pathways: Endothelial Cell FAK-A Target for Cancer Treatment.Clin Cancer Res. 2016 Aug 1;22(15):3718-24. doi: 10.1158/1078-0432.CCR-14-2021. Epub 2016 Jun 4. Clin Cancer Res. 2016. PMID: 27262114 Free PMC article. Review.
Cited by
-
OvCa-Chip microsystem recreates vascular endothelium-mediated platelet extravasation in ovarian cancer.Blood Adv. 2020 Jul 28;4(14):3329-3342. doi: 10.1182/bloodadvances.2020001632. Blood Adv. 2020. PMID: 32717032 Free PMC article.
-
Endothelial Cell Dysfunction Due to Molecules Secreted by Macrophages in Sepsis.Biomolecules. 2024 Aug 9;14(8):980. doi: 10.3390/biom14080980. Biomolecules. 2024. PMID: 39199368 Free PMC article. Review.
-
Dialogue between VE-Cadherin and Sphingosine 1 Phosphate Receptor1 (S1PR1) for Protecting Endothelial Functions.Int J Mol Sci. 2023 Feb 16;24(4):4018. doi: 10.3390/ijms24044018. Int J Mol Sci. 2023. PMID: 36835432 Free PMC article. Review.
-
Focal adhesion kinase (FAK) activation by estrogens involves GPER in triple-negative breast cancer cells.J Exp Clin Cancer Res. 2019 Feb 6;38(1):58. doi: 10.1186/s13046-019-1056-8. J Exp Clin Cancer Res. 2019. PMID: 30728047 Free PMC article.
-
Neuronal Wiskott-Aldrich syndrome protein regulates TGF-β1-mediated lung vascular permeability.FASEB J. 2016 Jul;30(7):2557-69. doi: 10.1096/fj.201600102R. Epub 2016 Mar 29. FASEB J. 2016. PMID: 27025963 Free PMC article.
References
-
- Abu-Ghazaleh R., Kabir J., Jia H., Lobo M., Zachary I. 2001. Src mediates stimulation by vascular endothelial growth factor of the phosphorylation of focal adhesion kinase at tyrosine 861, and migration and anti-apoptosis in endothelial cells. Biochem. J. 360:255–264 10.1042/0264-6021:3600255 - DOI - PMC - PubMed
-
- Allingham M.J., van Buul J.D., Burridge K. 2007. ICAM-1-mediated, Src- and Pyk2-dependent vascular endothelial cadherin tyrosine phosphorylation is required for leukocyte transendothelial migration. J. Immunol. 179:4053–4064 - PubMed
-
- Bobek V., Kolostova K., Pinterova D., Kacprzak G., Adamiak J., Kolodziej J., Boubelik M., Kubecova M., Hoffman R.M. 2010. A clinically relevant, syngeneic model of spontaneous, highly metastatic B16 mouse melanoma. Anticancer Res. 30:4799–4803 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- 200810MFE-193594-139144/CAPMC/ CIHR/Canada
- R37 CA050286/CA/NCI NIH HHS/United States
- R01 CA102310/CA/NCI NIH HHS/United States
- CA102310/CA/NCI NIH HHS/United States
- R01 CA180769/CA/NCI NIH HHS/United States
- R37 CA50286/CA/NCI NIH HHS/United States
- HL093156/HL/NHLBI NIH HHS/United States
- F32CA159558/CA/NCI NIH HHS/United States
- P30 CA023100/CA/NCI NIH HHS/United States
- F32 CA159558/CA/NCI NIH HHS/United States
- PG/11/62/29010/BHF_/British Heart Foundation/United Kingdom
- R01 HL093156/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
