

KRAS: feeding pancreatic cancer proliferation
- PMID: 24388967
- PMCID: PMC3955735
- DOI: 10.1016/j.tibs.2013.12.004
KRAS: feeding pancreatic cancer proliferation
Abstract
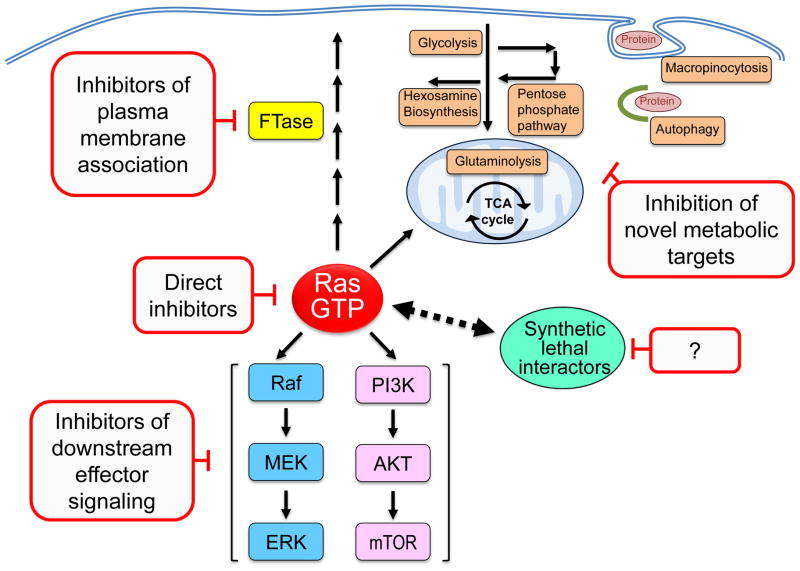
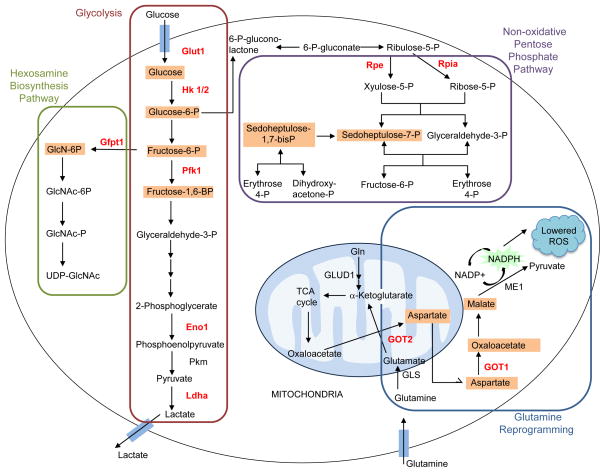
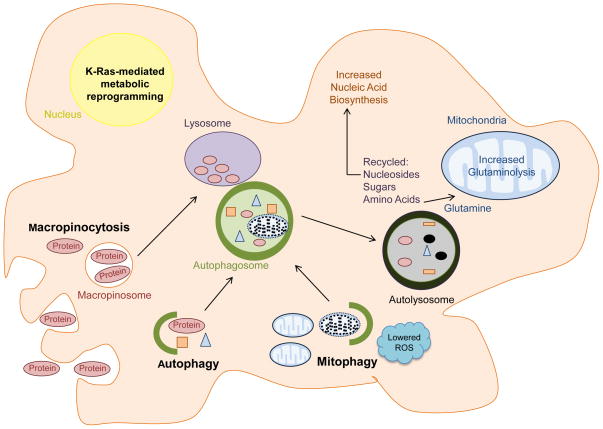
Oncogenic KRAS mutation is the signature genetic event in the progression and growth of pancreatic ductal adenocarcinoma (PDAC), an almost universally fatal disease. Although it has been appreciated for some time that nearly 95% of PDAC harbor mutationally activated KRAS, to date no effective treatments that target this mutant protein have reached the clinic. A number of studies have shown that oncogenic KRAS plays a central role in controlling tumor metabolism by orchestrating multiple metabolic changes including stimulation of glucose uptake, differential channeling of glucose intermediates, reprogrammed glutamine metabolism, increased autophagy, and macropinocytosis. We review these recent findings and address how they may be applied to develop new PDAC treatments.
Keywords: autophagy; glutaminolysis; glycolysis; macropinocytosis; metabolism.
Copyright © 2013 Elsevier Ltd. All rights reserved.
Figures
Similar articles
-
Ribonucleoprotein HNRNPA2B1 interacts with and regulates oncogenic KRAS in pancreatic ductal adenocarcinoma cells.Gastroenterology. 2014 Oct;147(4):882-892.e8. doi: 10.1053/j.gastro.2014.06.041. Epub 2014 Jul 3. Gastroenterology. 2014. PMID: 24998203
-
Paraoxonase 2 Facilitates Pancreatic Cancer Growth and Metastasis by Stimulating GLUT1-Mediated Glucose Transport.Mol Cell. 2017 Aug 17;67(4):685-701.e6. doi: 10.1016/j.molcel.2017.07.014. Epub 2017 Aug 10. Mol Cell. 2017. PMID: 28803777 Free PMC article.
-
Atypical KRASG12R Mutant Is Impaired in PI3K Signaling and Macropinocytosis in Pancreatic Cancer.Cancer Discov. 2020 Jan;10(1):104-123. doi: 10.1158/2159-8290.CD-19-1006. Epub 2019 Oct 24. Cancer Discov. 2020. PMID: 31649109 Free PMC article.
-
Oncogenic KRAS signalling in pancreatic cancer.Br J Cancer. 2014 Aug 26;111(5):817-22. doi: 10.1038/bjc.2014.215. Epub 2014 Apr 22. Br J Cancer. 2014. PMID: 24755884 Free PMC article. Review.
-
Epidemiological-molecular evidence of metabolic reprogramming on proliferation, autophagy and cell signaling in pancreas cancer.Cancer Lett. 2015 Jan 28;356(2 Pt A):281-8. doi: 10.1016/j.canlet.2014.03.028. Epub 2014 Apr 2. Cancer Lett. 2015. PMID: 24704294 Review.
Cited by
-
The BET bromodomain inhibitor JQ1 suppresses growth of pancreatic ductal adenocarcinoma in patient-derived xenograft models.Oncogene. 2016 Feb 18;35(7):833-45. doi: 10.1038/onc.2015.126. Epub 2015 May 11. Oncogene. 2016. PMID: 25961927 Free PMC article.
-
RAS Function in cancer cells: translating membrane biology and biochemistry into new therapeutics.Biochem J. 2020 Aug 14;477(15):2893-2919. doi: 10.1042/BCJ20190839. Biochem J. 2020. PMID: 32797215 Free PMC article. Review.
-
Orchestration of mesenchymal plasticity and immune evasiveness via rewiring of the metabolic program in pancreatic ductal adenocarcinoma.Front Oncol. 2022 Nov 3;12:1005566. doi: 10.3389/fonc.2022.1005566. eCollection 2022. Front Oncol. 2022. PMID: 36408139 Free PMC article. Review.
-
Pancreatic cancer mutationscape: revealing the link between modular restructuring and intervention efficacy amidst common mutations.bioRxiv [Preprint]. 2024 May 22:2024.01.27.577546. doi: 10.1101/2024.01.27.577546. bioRxiv. 2024. Update in: NPJ Syst Biol Appl. 2024 Jul 13;10(1):74. doi: 10.1038/s41540-024-00398-6. PMID: 38352601 Free PMC article. Updated. Preprint.
-
Integrating Genomics and Clinical Data for Statistical Analysis by Using GEnome MINIng (GEMINI) and Fast Healthcare Interoperability Resources (FHIR): System Design and Implementation.J Med Internet Res. 2020 Oct 7;22(10):e19879. doi: 10.2196/19879. J Med Internet Res. 2020. PMID: 33026356 Free PMC article.
References
-
- Scheffzek K, et al. The Ras-RasGAP complex: structural basis for GTPase activation and its loss in oncogenic Ras mutants. Science. 1997;277:333–338. - PubMed
-
- Scheidig AJ, et al. The pre-hydrolysis state of p21(ras) in complex with GTP: new insights into the role of water molecules in the GTP hydrolysis reaction of ras-like proteins. Structure. 1999;7:1311–1324. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
