

Extracellular regulation of VEGF: isoforms, proteolysis, and vascular patterning
- PMID: 24332926
- PMCID: PMC3977708
- DOI: 10.1016/j.cytogfr.2013.11.002
Extracellular regulation of VEGF: isoforms, proteolysis, and vascular patterning
Abstract
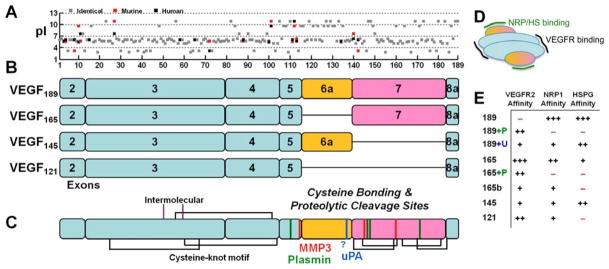
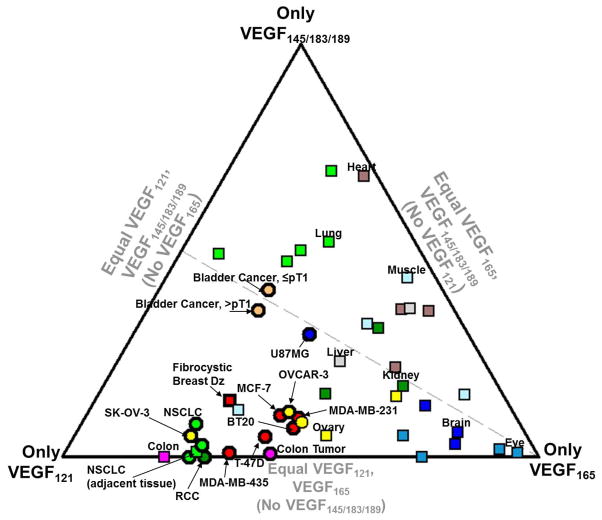
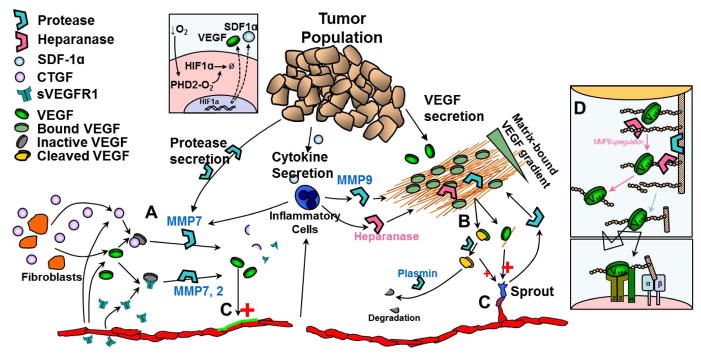
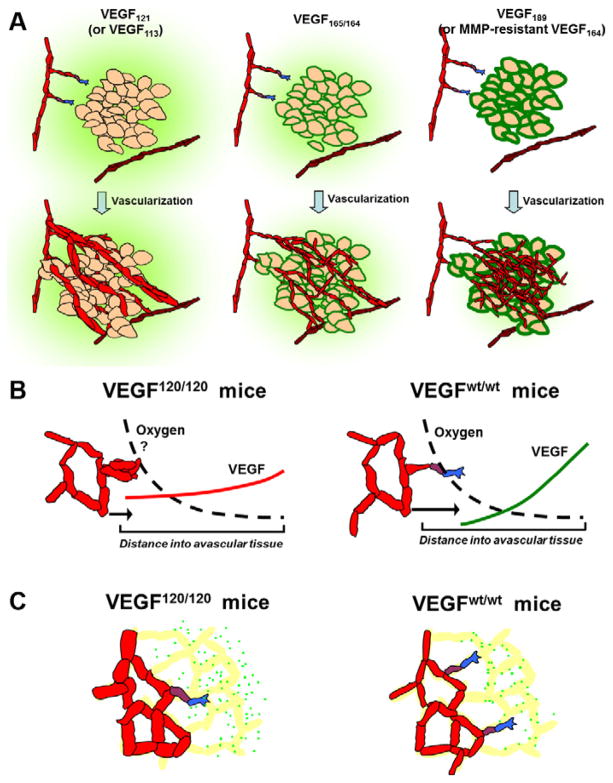
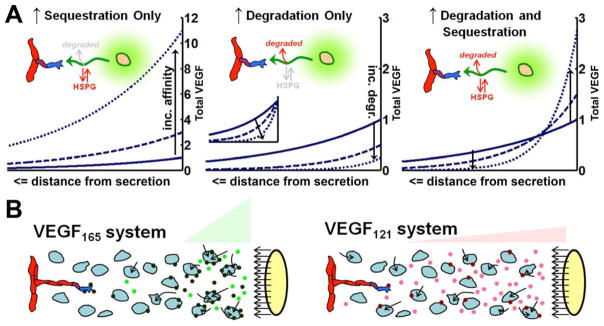
The regulation of vascular endothelial growth factor A (VEGF) is critical to neovascularization in numerous tissues under physiological and pathological conditions. VEGF has multiple isoforms, created by alternative splicing or proteolytic cleavage, and characterized by different receptor-binding and matrix-binding properties. These isoforms are known to give rise to a spectrum of angiogenesis patterns marked by differences in branching, which has functional implications for tissues. In this review, we detail the extensive extracellular regulation of VEGF and the ability of VEGF to dictate the vascular phenotype. We explore the role of VEGF-releasing proteases and soluble carrier molecules on VEGF activity. While proteases such as MMP9 can 'release' matrix-bound VEGF and promote angiogenesis, for example as a key step in carcinogenesis, proteases can also suppress VEGF's angiogenic effects. We explore what dictates pro- or anti-angiogenic behavior. We also seek to understand the phenomenon of VEGF gradient formation. Strong VEGF gradients are thought to be due to decreased rates of diffusion from reversible matrix binding, however theoretical studies show that this scenario cannot give rise to lasting VEGF gradients in vivo. We propose that gradients are formed through degradation of sequestered VEGF. Finally, we review how different aspects of the VEGF signal, such as its concentration, gradient, matrix-binding, and NRP1-binding can differentially affect angiogenesis. We explore how this allows VEGF to regulate the formation of vascular networks across a spectrum of high to low branching densities, and from normal to pathological angiogenesis. A better understanding of the control of angiogenesis is necessary to improve upon limitations of current angiogenic therapies.
Keywords: Angiogenesis; Computational model; Extracellular matrix; Gradient; Mathematical model; Microenvironment; Protease; Receptor; Systems biology.
Copyright © 2013 Elsevier Ltd. All rights reserved.
Figures
Similar articles
-
Formation of VEGF isoform-specific spatial distributions governing angiogenesis: computational analysis.BMC Syst Biol. 2011 May 2;5:59. doi: 10.1186/1752-0509-5-59. BMC Syst Biol. 2011. PMID: 21535871 Free PMC article.
-
Spatial distribution of VEGF isoforms and chemotactic signals in the vicinity of a tumor.J Theor Biol. 2008 Jun 21;252(4):593-607. doi: 10.1016/j.jtbi.2008.02.009. Epub 2008 Feb 16. J Theor Biol. 2008. PMID: 18395755
-
VEGF (Vascular Endothelial Growth Factor) Induces NRP1 (Neuropilin-1) Cleavage via ADAMs (a Disintegrin and Metalloproteinase) 9 and 10 to Generate Novel Carboxy-Terminal NRP1 Fragments That Regulate Angiogenic Signaling.Arterioscler Thromb Vasc Biol. 2018 Aug;38(8):1845-1858. doi: 10.1161/ATVBAHA.118.311118. Arterioscler Thromb Vasc Biol. 2018. PMID: 29880492 Free PMC article.
-
Binding to the extracellular matrix and proteolytic processing: two key mechanisms regulating vascular endothelial growth factor action.Mol Biol Cell. 2010 Mar 1;21(5):687-90. doi: 10.1091/mbc.e09-07-0590. Mol Biol Cell. 2010. PMID: 20185770 Free PMC article. Review.
-
Role of the vascular endothelial growth factor isoforms in retinal angiogenesis and DiGeorge syndrome.Verh K Acad Geneeskd Belg. 2005;67(4):229-76. Verh K Acad Geneeskd Belg. 2005. PMID: 16334858 Review.
Cited by
-
Bioprinted vascular tissue: Assessing functions from cellular, tissue to organ levels.Mater Today Bio. 2023 Oct 28;23:100846. doi: 10.1016/j.mtbio.2023.100846. eCollection 2023 Dec. Mater Today Bio. 2023. PMID: 37953757 Free PMC article. Review.
-
Circular RNA CircSHKBP1 accelerates the proliferation, invasion, angiogenesis, and stem cell-like properties via modulation of microR-766-5p/high mobility group AT-hook 2 axis in laryngeal squamous cell carcinoma.Bioengineered. 2022 May;13(5):11551-11563. doi: 10.1080/21655979.2022.2068922. Bioengineered. 2022. PMID: 35502885 Free PMC article.
-
Associations of microRNAs, Angiogenesis-Regulating Factors and CFH Y402H Polymorphism-An Attempt to Search for Systemic Biomarkers in Age-Related Macular Degeneration.Int J Mol Sci. 2019 Nov 15;20(22):5750. doi: 10.3390/ijms20225750. Int J Mol Sci. 2019. PMID: 31731799 Free PMC article.
-
Human Umbilical Tissue-Derived Cells Secrete Soluble VEGFR1 and Inhibit Choroidal Neovascularization.Mol Ther Methods Clin Dev. 2019 May 22;14:37-46. doi: 10.1016/j.omtm.2019.05.007. eCollection 2019 Sep 13. Mol Ther Methods Clin Dev. 2019. PMID: 31276010 Free PMC article.
-
Proteases Regulate Cancer Stem Cell Properties and Remodel Their Microenvironment.J Histochem Cytochem. 2021 Dec;69(12):775-794. doi: 10.1369/00221554211035192. Epub 2021 Jul 26. J Histochem Cytochem. 2021. PMID: 34310223 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
