

Genetically encoded fluorescent probe to visualize intracellular phosphatidylinositol 3,5-bisphosphate localization and dynamics
- PMID: 24324172
- PMCID: PMC3876232
- DOI: 10.1073/pnas.1311864110
Genetically encoded fluorescent probe to visualize intracellular phosphatidylinositol 3,5-bisphosphate localization and dynamics
Abstract
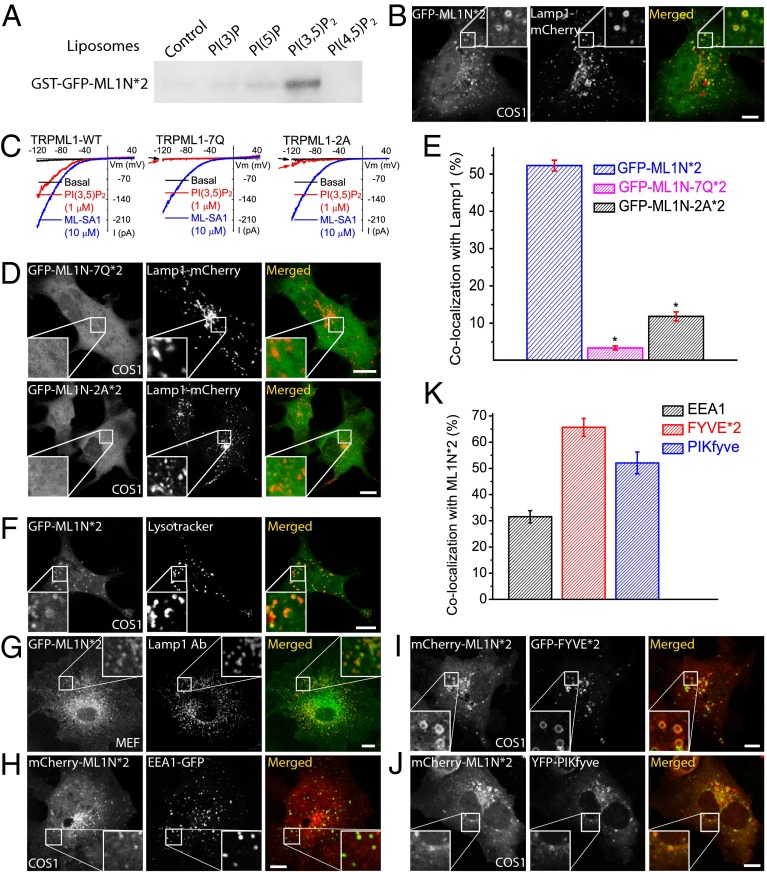
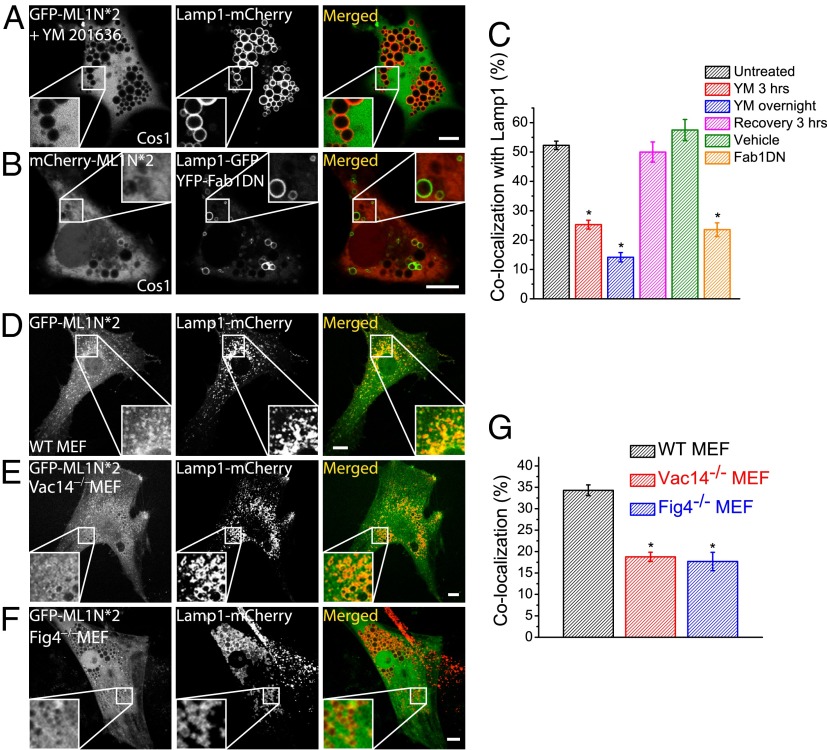
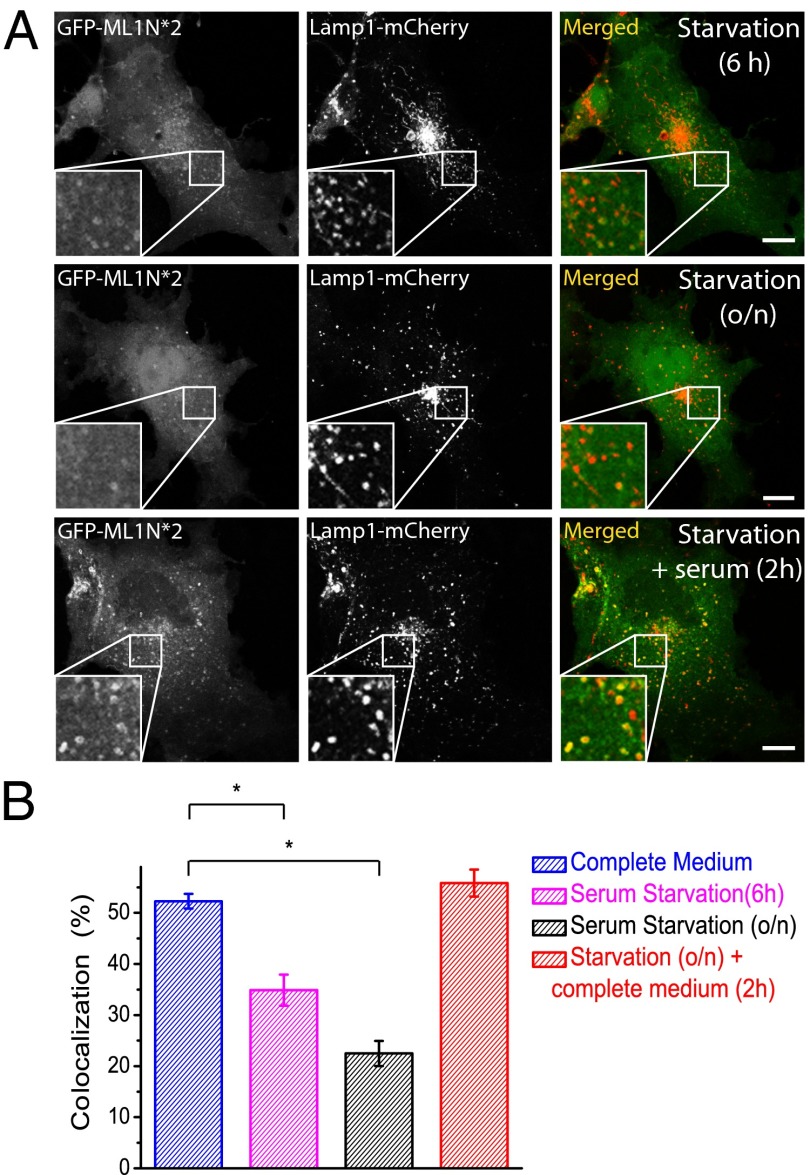
Phosphatidylinositol 3,5-bisphosphate [PI(3,5)P2] is a low-abundance phosphoinositide presumed to be localized to endosomes and lysosomes, where it recruits cytoplasmic peripheral proteins and regulates endolysosome-localized membrane channel activity. Cells lacking PI(3,5)P2 exhibit lysosomal trafficking defects, and human mutations in the PI(3,5)P2-metabolizing enzymes cause lysosome-related diseases. The spatial and temporal dynamics of PI(3,5)P2, however, remain unclear due to the lack of a reliable detection method. Of the seven known phosphoinositides, only PI(3,5)P2 binds, in the low nanomolar range, to a cytoplasmic phosphoinositide-interacting domain (ML1N) to activate late endosome and lysosome (LEL)-localized transient receptor potential Mucolipin 1 (TRPML1) channels. Here, we report the generation and characterization of a PI(3,5)P2-specific probe, generated by the fusion of fluorescence tags to the tandem repeats of ML1N. The probe was mainly localized to the membranes of Lamp1-positive compartments, and the localization pattern was dynamically altered by either mutations in the probe, or by genetically or pharmacologically manipulating the cellular levels of PI(3,5)P2. Through the use of time-lapse live-cell imaging, we found that the localization of the PI(3,5)P2 probe was regulated by serum withdrawal/addition, undergoing rapid changes immediately before membrane fusion of two LELs. Our development of a PI(3,5)P2-specific probe may facilitate studies of both intracellular signal transduction and membrane trafficking in the endosomes and lysosomes.
Keywords: PIKfyve; TRP channel; vesicle fusion.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

Similar articles
-
Visualization of Phosphatidylinositol 3,5-Bisphosphate Dynamics by a Tandem ML1N-Based Fluorescent Protein Probe in Arabidopsis.Plant Cell Physiol. 2017 Jul 1;58(7):1185-1195. doi: 10.1093/pcp/pcx011. Plant Cell Physiol. 2017. PMID: 28158631 Free PMC article.
-
Drosophila TRPML forms PI(3,5)P2-activated cation channels in both endolysosomes and plasma membrane.J Biol Chem. 2014 Feb 14;289(7):4262-72. doi: 10.1074/jbc.M113.506501. Epub 2013 Dec 27. J Biol Chem. 2014. PMID: 24375408 Free PMC article.
-
PI(3,5)P(2) controls membrane trafficking by direct activation of mucolipin Ca(2+) release channels in the endolysosome.Nat Commun. 2010 Jul 13;1(4):38. doi: 10.1038/ncomms1037. Nat Commun. 2010. PMID: 20802798 Free PMC article.
-
TRPML1: an ion channel in the lysosome.Handb Exp Pharmacol. 2014;222:631-45. doi: 10.1007/978-3-642-54215-2_24. Handb Exp Pharmacol. 2014. PMID: 24756723 Review.
-
TRPML2 and mucolipin evolution.Handb Exp Pharmacol. 2014;222:647-58. doi: 10.1007/978-3-642-54215-2_25. Handb Exp Pharmacol. 2014. PMID: 24756724 Review.
Cited by
-
Targeting cancer metabolism by simultaneously disrupting parallel nutrient access pathways.J Clin Invest. 2016 Nov 1;126(11):4088-4102. doi: 10.1172/JCI87148. Epub 2016 Sep 26. J Clin Invest. 2016. PMID: 27669461 Free PMC article.
-
Cholesterol transfer via endoplasmic reticulum contacts mediates lysosome damage repair.EMBO J. 2022 Dec 15;41(24):e112677. doi: 10.15252/embj.2022112677. Epub 2022 Nov 21. EMBO J. 2022. PMID: 36408828 Free PMC article.
-
Lipid-gated monovalent ion fluxes regulate endocytic traffic and support immune surveillance.Science. 2020 Jan 17;367(6475):301-305. doi: 10.1126/science.aaw9544. Epub 2019 Dec 5. Science. 2020. PMID: 31806695 Free PMC article.
-
An update on genetically encoded lipid biosensors.Mol Biol Cell. 2022 May 1;33(5):tp2. doi: 10.1091/mbc.E21-07-0363. Mol Biol Cell. 2022. PMID: 35420888 Free PMC article.
-
Diverse Physiological Functions of FAB1 and Phosphatidylinositol 3,5-Bisphosphate in Plants.Front Plant Sci. 2019 Mar 22;10:274. doi: 10.3389/fpls.2019.00274. eCollection 2019. Front Plant Sci. 2019. PMID: 30967882 Free PMC article. Review.
References
-
- Di Paolo G, De Camilli P. Phosphoinositides in cell regulation and membrane dynamics. Nature. 2006;443(7112):651–657. - PubMed
-
- Lemmon MA. Membrane recognition by phospholipid-binding domains. Nat Rev Mol Cell Biol. 2008;9(2):99–111. - PubMed
-
- Stauffer TP, Ahn S, Meyer T. Receptor-induced transient reduction in plasma membrane PtdIns(4,5)P2 concentration monitored in living cells. Curr Biol. 1998;8(6):343–346. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
