

Genetic variation of a bacterial pathogen within individuals with cystic fibrosis provides a record of selective pressures
- PMID: 24316980
- PMCID: PMC3979468
- DOI: 10.1038/ng.2848
Genetic variation of a bacterial pathogen within individuals with cystic fibrosis provides a record of selective pressures
Abstract
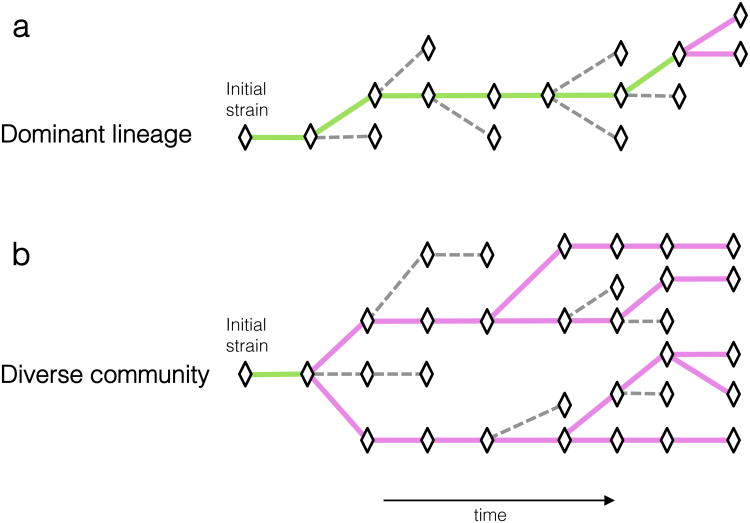
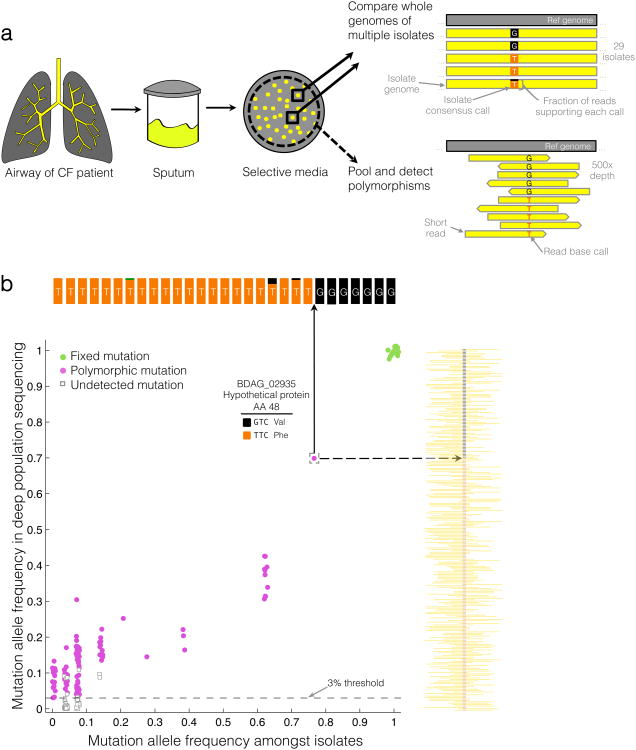
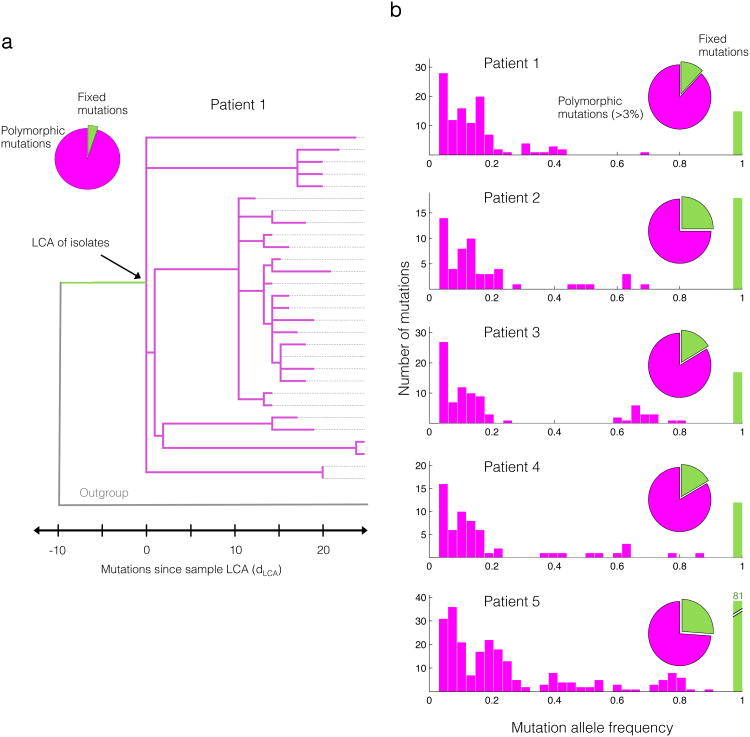
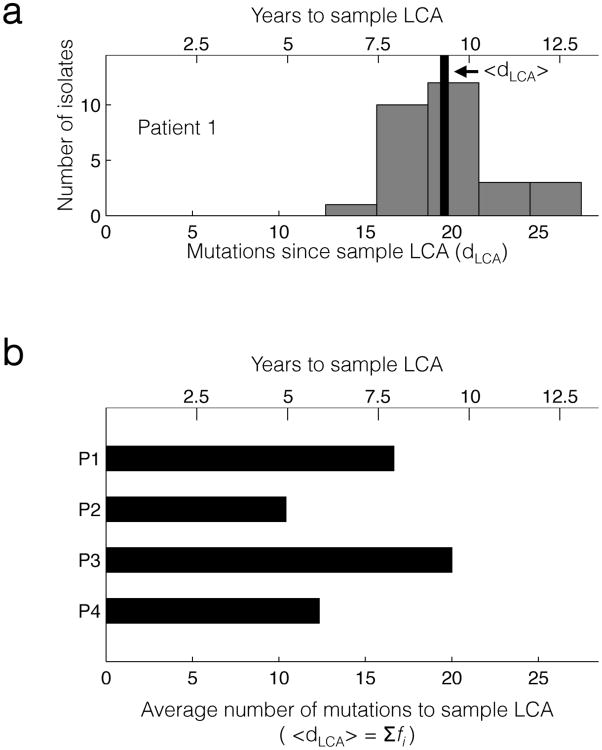
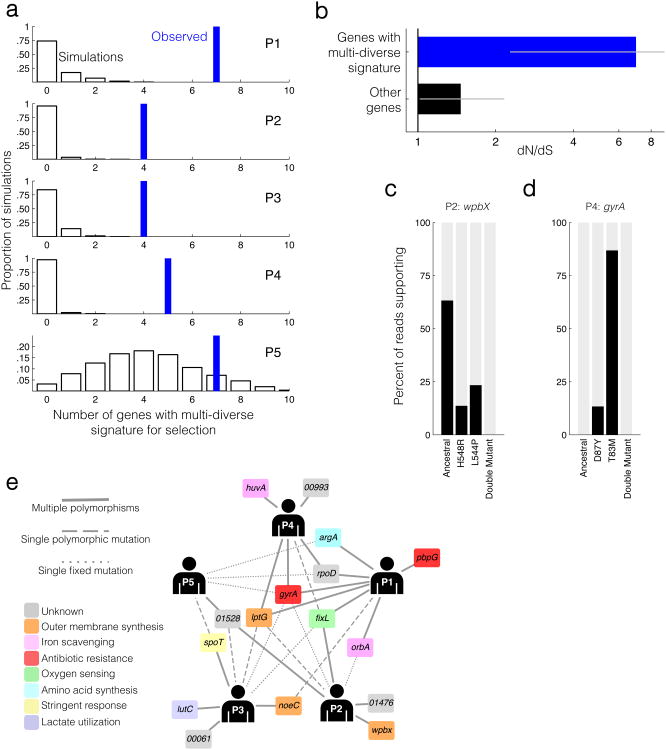
Advances in sequencing technologies have enabled the identification of mutations acquired by bacterial pathogens during infection. However, it remains unclear whether adaptive mutations fix in the population or lead to pathogen diversification within the patient. Here we study the genotypic diversity of Burkholderia dolosa within individuals with cystic fibrosis by resequencing individual colonies and whole populations from single sputum samples. We find extensive intrasample diversity, suggesting that mutations rarely fix in a patient's pathogen population--instead, diversifying lineages coexist for many years. Under strong selection, multiple adaptive mutations arise, but none of these sweep to fixation, generating lasting allele diversity that provides a recorded signature of past selection. Genes involved in outer-membrane components, iron scavenging and antibiotic resistance all showed this signature of within-patient selection. These results offer a general and rapid approach for identifying the selective pressures acting on a pathogen in individual patients based on single clinical samples.
Figures
Comment in
-
Genetic signature of bacterial pathogen adaptation during chronic pulmonary infections.Nat Genet. 2014 Jan;46(1):5-6. doi: 10.1038/ng.2859. Nat Genet. 2014. PMID: 24370741
Similar articles
-
Genetic signature of bacterial pathogen adaptation during chronic pulmonary infections.Nat Genet. 2014 Jan;46(1):5-6. doi: 10.1038/ng.2859. Nat Genet. 2014. PMID: 24370741
-
Parallel bacterial evolution within multiple patients identifies candidate pathogenicity genes.Nat Genet. 2011 Nov 13;43(12):1275-80. doi: 10.1038/ng.997. Nat Genet. 2011. PMID: 22081229 Free PMC article.
-
Long-term colonization of the cystic fibrosis lung by Burkholderia cepacia complex bacteria: epidemiology, clonal variation, and genome-wide expression alterations.Front Cell Infect Microbiol. 2011 Dec 2;1:12. doi: 10.3389/fcimb.2011.00012. eCollection 2011. Front Cell Infect Microbiol. 2011. PMID: 22919578 Free PMC article.
-
Insights into the role of extracellular polysaccharides in Burkholderia adaptation to different environments.Front Cell Infect Microbiol. 2011 Dec 15;1:16. doi: 10.3389/fcimb.2011.00016. eCollection 2011. Front Cell Infect Microbiol. 2011. PMID: 22919582 Free PMC article. Review.
-
Burkholderia cenocepacia in cystic fibrosis: epidemiology and molecular mechanisms of virulence.Clin Microbiol Infect. 2010 Jul;16(7):821-30. doi: 10.1111/j.1469-0691.2010.03237.x. Clin Microbiol Infect. 2010. PMID: 20880411 Review.
Cited by
-
A genome-wide association analysis reveals a potential role for recombination in the evolution of antimicrobial resistance in Burkholderia multivorans.PLoS Pathog. 2018 Dec 7;14(12):e1007453. doi: 10.1371/journal.ppat.1007453. eCollection 2018 Dec. PLoS Pathog. 2018. PMID: 30532201 Free PMC article.
-
Population dynamics of the human gut microbiome: change is the only constant.Genome Biol. 2019 Jul 31;20(1):150. doi: 10.1186/s13059-019-1775-3. Genome Biol. 2019. PMID: 31366367 Free PMC article.
-
Single-cell genomics of co-sorted Nanoarchaeota suggests novel putative host associations and diversification of proteins involved in symbiosis.Microbiome. 2018 Sep 17;6(1):161. doi: 10.1186/s40168-018-0539-8. Microbiome. 2018. PMID: 30223889 Free PMC article.
-
Anatomy promotes neutral coexistence of strains in the human skin microbiome.Cell Host Microbe. 2022 Feb 9;30(2):171-182.e7. doi: 10.1016/j.chom.2021.12.007. Epub 2022 Jan 6. Cell Host Microbe. 2022. PMID: 34995483 Free PMC article.
-
Rapid methicillin resistance diversification in Staphylococcus epidermidis colonizing human neonates.Nat Commun. 2021 Oct 18;12(1):6062. doi: 10.1038/s41467-021-26392-8. Nat Commun. 2021. PMID: 34663826 Free PMC article.
References
Publication types
MeSH terms
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
