

Early gene expression changes in spinal cord from SOD1(G93A) Amyotrophic Lateral Sclerosis animal model
- PMID: 24302897
- PMCID: PMC3831149
- DOI: 10.3389/fncel.2013.00216
Early gene expression changes in spinal cord from SOD1(G93A) Amyotrophic Lateral Sclerosis animal model
Abstract
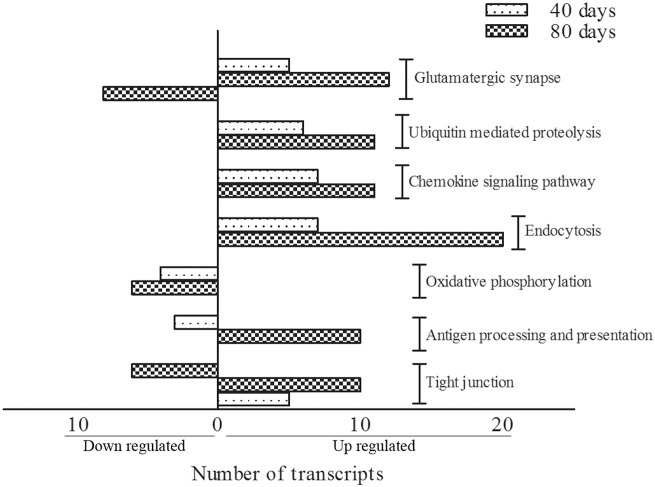
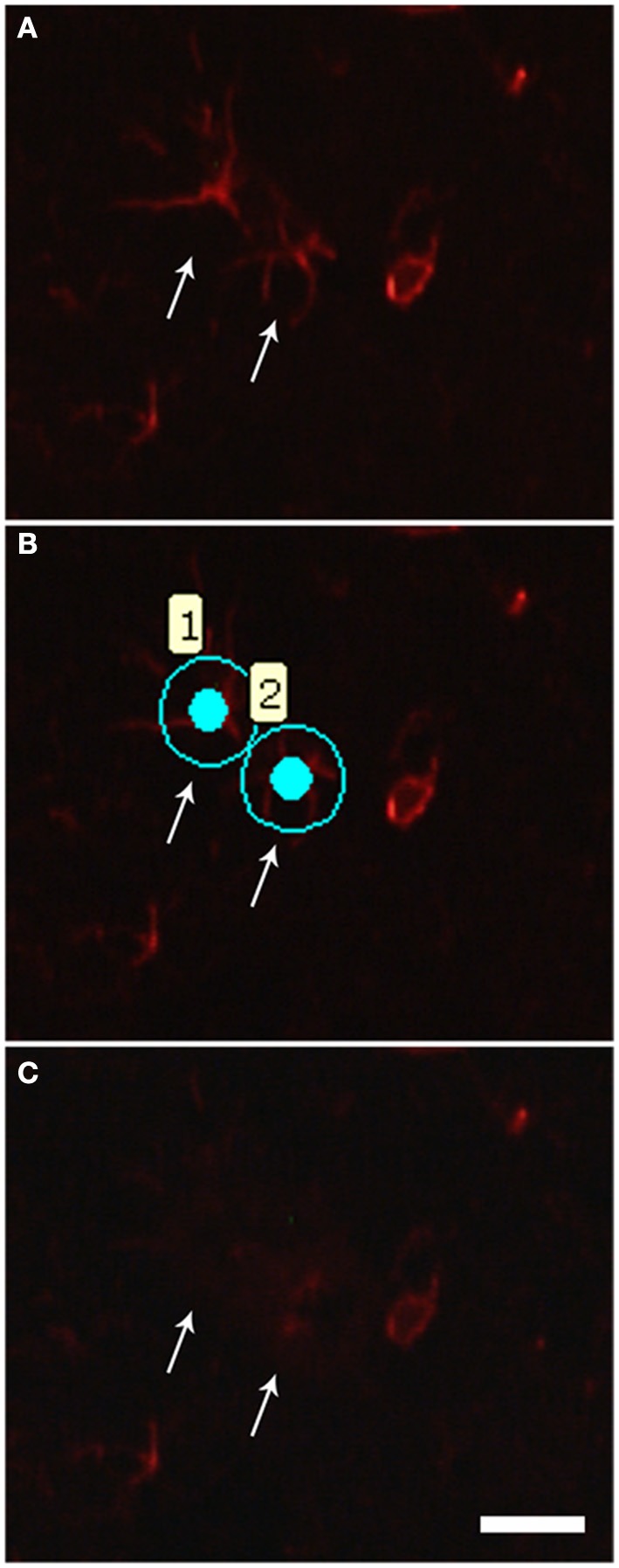
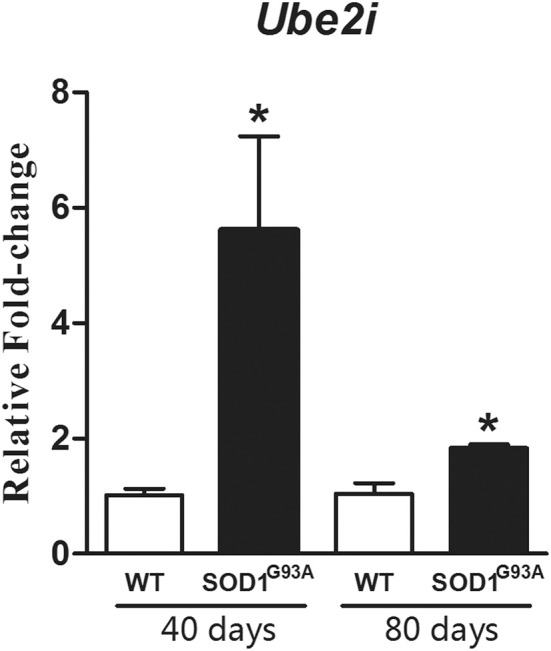
Amyotrophic Lateral Sclerosis (ALS) is an adult-onset and fast progression neurodegenerative disease that leads to the loss of motor neurons. Mechanisms of selective motor neuron loss in ALS are unknown. The early events occurring in the spinal cord that may contribute to motor neuron death are not described, neither astrocytes participation in the pre-symptomatic phases of the disease. In order to identify ALS early events, we performed a microarray analysis employing a whole mouse genome platform to evaluate the gene expression pattern of lumbar spinal cords of transgenic SOD1(G93A) mice and their littermate controls at pre-symptomatic ages of 40 and 80 days. Differentially expressed genes were identified by means of the Bioconductor packages Agi4×44Preprocess and limma. FunNet web based tool was used for analysis of over-represented pathways. Furthermore, immunolabeled astrocytes from 40 and 80 days old mice were submitted to laser microdissection and RNA was extracted for evaluation of a selected gene by qPCR. Statistical analysis has pointed to 492 differentially expressed genes (155 up and 337 down regulated) in 40 days and 1105 (433 up and 672 down) in 80 days old ALS mice. KEGG analysis demonstrated the over-represented pathways tight junction, antigen processing and presentation, oxidative phosphorylation, endocytosis, chemokine signaling pathway, ubiquitin mediated proteolysis and glutamatergic synapse at both pre-symptomatic ages. Ube2i gene expression was evaluated in astrocytes from both transgenic ages, being up regulated in 40 and 80 days astrocytes enriched samples. Our data points to important early molecular events occurring in pre-symptomatic phases of ALS in mouse model. Early SUMOylation process linked to astrocytes might account to non-autonomous cell toxicity in ALS. Further studies on the signaling pathways presented here may provide new insights to better understand the events triggering motor neuron death in this devastating disorder.
Keywords: ALS; SOD1G93A; astrocytes; laser microdissection; microarray; pre-symptomatic; spinal cord.
Figures
Similar articles
-
Deregulated expression of cytoskeleton related genes in the spinal cord and sciatic nerve of presymptomatic SOD1(G93A) Amyotrophic Lateral Sclerosis mouse model.Front Cell Neurosci. 2014 May 26;8:148. doi: 10.3389/fncel.2014.00148. eCollection 2014. Front Cell Neurosci. 2014. PMID: 24904291 Free PMC article.
-
Toll-Like Receptor-4 Inhibitor TAK-242 Attenuates Motor Dysfunction and Spinal Cord Pathology in an Amyotrophic Lateral Sclerosis Mouse Model.Int J Mol Sci. 2017 Aug 1;18(8):1666. doi: 10.3390/ijms18081666. Int J Mol Sci. 2017. PMID: 28763002 Free PMC article.
-
Lysosomal and phagocytic activity is increased in astrocytes during disease progression in the SOD1 (G93A) mouse model of amyotrophic lateral sclerosis.Front Cell Neurosci. 2015 Oct 15;9:410. doi: 10.3389/fncel.2015.00410. eCollection 2015. Front Cell Neurosci. 2015. PMID: 26528138 Free PMC article.
-
Altered expression of atypical PKC and Ryk in the spinal cord of a mouse model of amyotrophic lateral sclerosis.Dev Neurobiol. 2014 Aug;74(8):839-50. doi: 10.1002/dneu.22137. Epub 2014 Jan 22. Dev Neurobiol. 2014. PMID: 24123880 Free PMC article. Review.
-
Astrocytes and Microglia as Non-cell Autonomous Players in the Pathogenesis of ALS.Exp Neurobiol. 2016 Oct;25(5):233-240. doi: 10.5607/en.2016.25.5.233. Epub 2016 Oct 20. Exp Neurobiol. 2016. PMID: 27790057 Free PMC article. Review.
Cited by
-
Arginine Methyltransferase PRMT8 Provides Cellular Stress Tolerance in Aging Motoneurons.J Neurosci. 2018 Aug 29;38(35):7683-7700. doi: 10.1523/JNEUROSCI.3389-17.2018. Epub 2018 Jul 27. J Neurosci. 2018. PMID: 30054395 Free PMC article.
-
Ultra-High Field Diffusion MRI Reveals Early Axonal Pathology in Spinal Cord of ALS mice.Transl Neurodegener. 2018 Aug 8;7:20. doi: 10.1186/s40035-018-0122-z. eCollection 2018. Transl Neurodegener. 2018. PMID: 30128146 Free PMC article.
-
Implications of genomic signatures in the differential vulnerability to fetal alcohol exposure in C57BL/6 and DBA/2 mice.Front Genet. 2014 Jun 11;5:173. doi: 10.3389/fgene.2014.00173. eCollection 2014. Front Genet. 2014. PMID: 24966868 Free PMC article.
-
ALS-Linked SOD1 Mutants Enhance Neurite Outgrowth and Branching in Adult Motor Neurons.iScience. 2019 Jan 25;11:294-304. doi: 10.1016/j.isci.2018.12.026. Epub 2018 Dec 27. iScience. 2019. PMID: 30639851 Free PMC article.
-
Correlating serum micrornas and clinical parameters in amyotrophic lateral sclerosis.Muscle Nerve. 2018 Aug;58(2):261-269. doi: 10.1002/mus.26106. Epub 2018 Mar 25. Muscle Nerve. 2018. PMID: 29466830 Free PMC article.
References
-
- Alves C. J., De Santana L. P., Dos Santos A. J., De Oliveira G. P., Duobles T., Scorisa J. M., et al. (2011). Early motor and electrophysiological changes in transgenic mouse model of amyotrophic lateral sclerosis and gender differences on clinical outcome. Brain Res. 1394, 90–104 10.1016/j.brainres.2011.02.060 - DOI - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous