

NRF2-regulation in brain health and disease: implication of cerebral inflammation
- PMID: 24262633
- PMCID: PMC3958930
- DOI: 10.1016/j.neuropharm.2013.11.004
NRF2-regulation in brain health and disease: implication of cerebral inflammation
Abstract
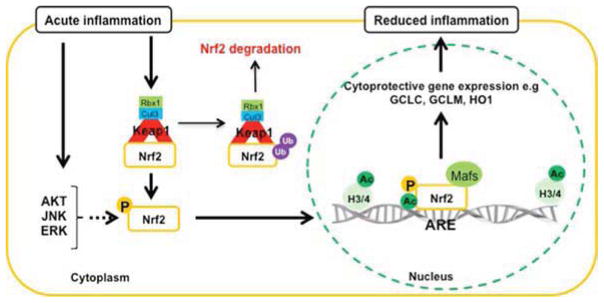
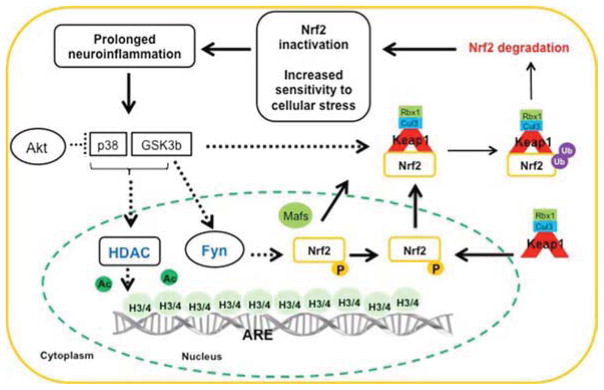
The nuclear factor erythroid 2 related factor 2 (NRF2) is a key regulator of endogenous inducible defense systems in the body. Under physiological conditions NRF2 is mainly located in the cytoplasm. However, in response to oxidative stress, NRF2 translocates to the nucleus and binds to specific DNA sites termed "anti-oxidant response elements" or "electrophile response elements" to initiate transcription of cytoprotective genes. Acute oxidative stress to the brain, such as stroke and traumatic brain injury is increased in animals that are deficient in NRF2. Insufficient NRF2 activation in humans has been linked to chronic diseases such as Parkinson's disease, Alzheimer's disease and amyotrophic lateral sclerosis. New findings have also linked activation of the NRF2 system to anti-inflammatory effects via interactions with NF-κB. Here we review literature on cellular mechanisms of NRF2 regulation, how to maintain and restore NRF2 function and the relationship between NRF2 regulation and brain damage. We bring forward the hypothesis that inflammation via prolonged activation of key kinases (p38 and GSK-3β) and activation of histone deacetylases gives rise to dysregulation of the NRF2 system in the brain, which contributes to oxidative stress and injury.
Keywords: Anti-oxidants; NRF2; Neuroinflammation.
Copyright © 2013 Elsevier Ltd. All rights reserved.
Figures
Similar articles
-
Icariin targets Nrf2 signaling to inhibit microglia-mediated neuroinflammation.Int Immunopharmacol. 2019 Aug;73:304-311. doi: 10.1016/j.intimp.2019.05.033. Epub 2019 May 22. Int Immunopharmacol. 2019. PMID: 31128530
-
Traumatic brain injury-induced downregulation of Nrf2 activates inflammatory response and apoptotic cell death.J Mol Med (Berl). 2019 Dec;97(12):1627-1641. doi: 10.1007/s00109-019-01851-4. Epub 2019 Nov 22. J Mol Med (Berl). 2019. PMID: 31758217
-
Counteracting role of nuclear factor erythroid 2-related factor 2 pathway in Alzheimer's disease.Biomed Pharmacother. 2020 Sep;129:110373. doi: 10.1016/j.biopha.2020.110373. Epub 2020 Jun 27. Biomed Pharmacother. 2020. PMID: 32603894 Review.
-
NRF2 and NF-қB interplay in cerebrovascular and neurodegenerative disorders: Molecular mechanisms and possible therapeutic approaches.Redox Biol. 2019 Feb;21:101059. doi: 10.1016/j.redox.2018.11.017. Epub 2018 Nov 28. Redox Biol. 2019. PMID: 30576920 Free PMC article. Review.
-
Synergistic Interaction Between Heme Oxygenase (HO) and Nuclear-Factor E2- Related Factor-2 (Nrf2) against Oxidative Stress in Cardiovascular Related Diseases.Curr Pharm Des. 2017;23(10):1465-1470. doi: 10.2174/1381612823666170113153818. Curr Pharm Des. 2017. PMID: 28088909 Review.
Cited by
-
Dietary Phytochemicals in Neuroimmunoaging: A New Therapeutic Possibility for Humans?Front Pharmacol. 2016 Oct 13;7:364. doi: 10.3389/fphar.2016.00364. eCollection 2016. Front Pharmacol. 2016. PMID: 27790141 Free PMC article. Review.
-
Marine Natural Products from the Russian Pacific as Sources of Drugs for Neurodegenerative Diseases.Mar Drugs. 2022 Nov 11;20(11):708. doi: 10.3390/md20110708. Mar Drugs. 2022. PMID: 36421986 Free PMC article. Review.
-
Protective Role of Epigallocatechin Gallate in a Rat Model of Cisplatin-Induced Cerebral Inflammation and Oxidative Damage: Impact of Modulating NF-κB and Nrf2.Neurotox Res. 2020 Feb;37(2):380-396. doi: 10.1007/s12640-019-00095-x. Epub 2019 Aug 13. Neurotox Res. 2020. PMID: 31410684
-
Ginsenoside Rg1 protects against ischemic/reperfusion-induced neuronal injury through miR-144/Nrf2/ARE pathway.Acta Pharmacol Sin. 2019 Jan;40(1):13-25. doi: 10.1038/s41401-018-0154-z. Epub 2018 Sep 27. Acta Pharmacol Sin. 2019. PMID: 30262824 Free PMC article.
-
Altered behavioral development in Nrf2 knockout mice following early postnatal exposure to valproic acid.Brain Res Bull. 2014 Oct;109:132-42. doi: 10.1016/j.brainresbull.2014.10.006. Epub 2014 Oct 20. Brain Res Bull. 2014. PMID: 25454122 Free PMC article.
References
-
- Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997;236:313–322. - PubMed
-
- Venugopal R, Jaiswal AK. Nrf2 and Nrf1 in association with Jun proteins regulate antioxidant response element-mediated expression and coordinated induction of genes encoding detoxifying enzymes. Oncogene. 1998;17:3145–3156. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
