

mTOR inhibition alleviates mitochondrial disease in a mouse model of Leigh syndrome
- PMID: 24231806
- PMCID: PMC4055856
- DOI: 10.1126/science.1244360
mTOR inhibition alleviates mitochondrial disease in a mouse model of Leigh syndrome
Abstract
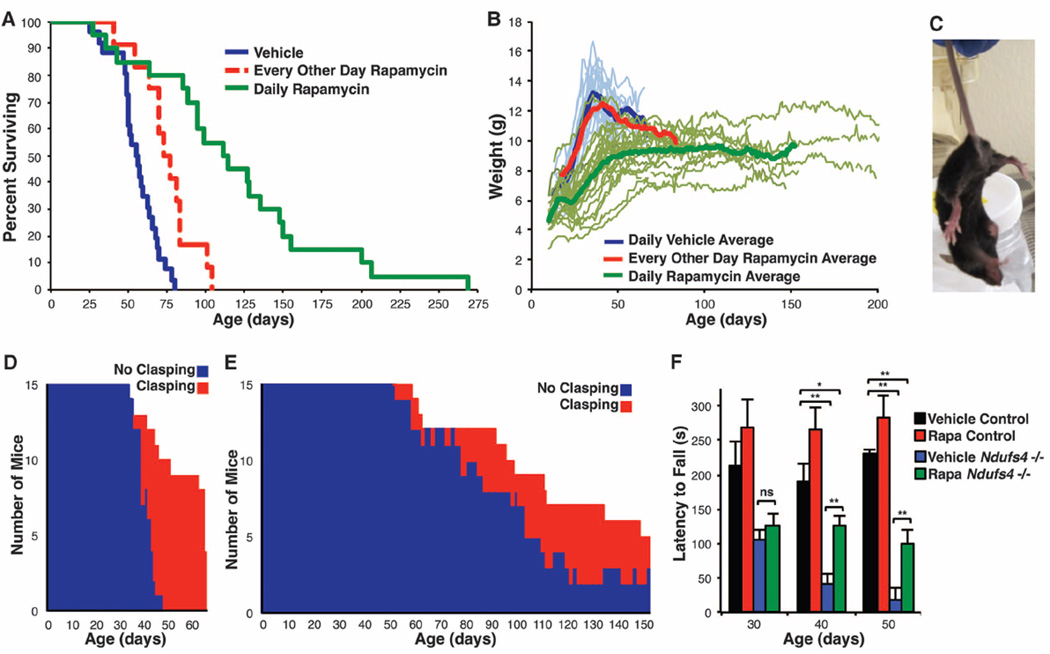
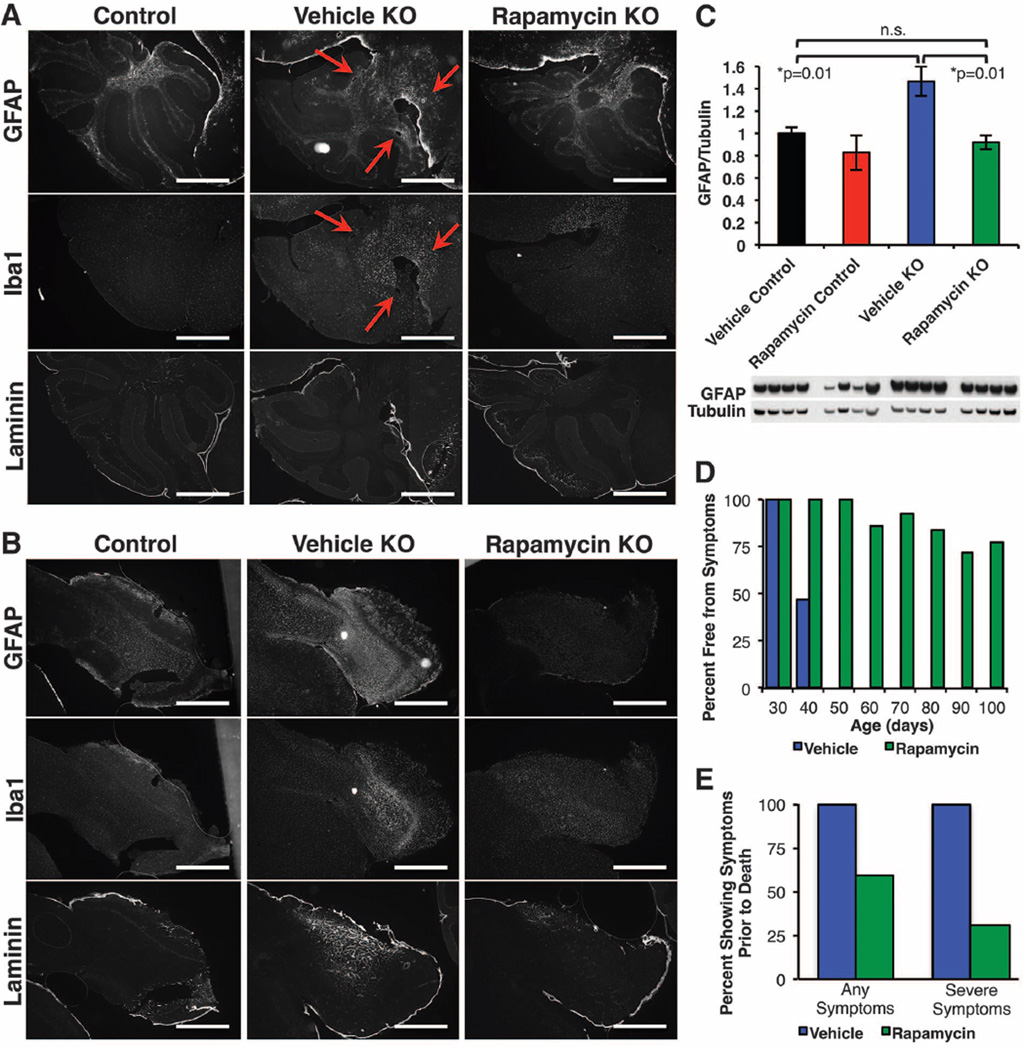
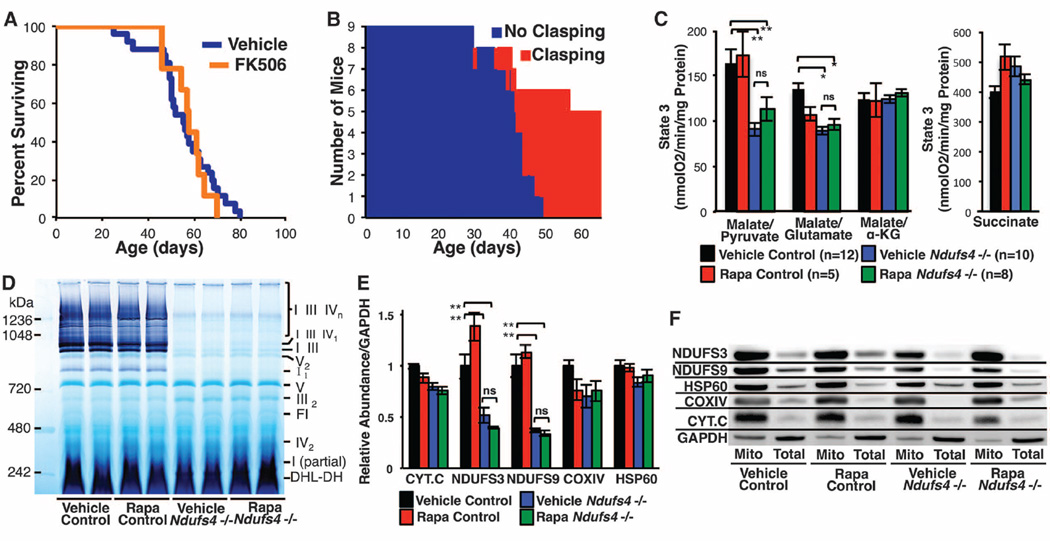
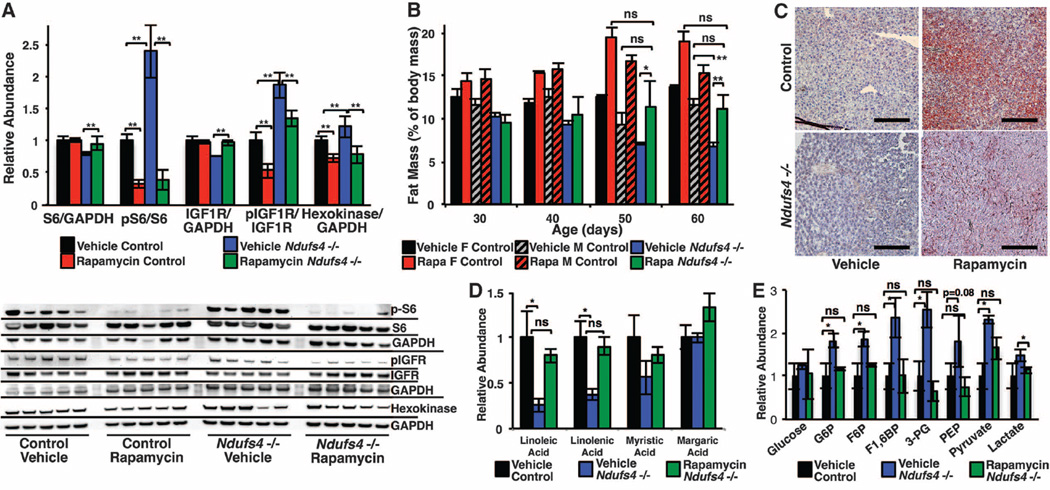
Mitochondrial dysfunction contributes to numerous health problems, including neurological and muscular degeneration, cardiomyopathies, cancer, diabetes, and pathologies of aging. Severe mitochondrial defects can result in childhood disorders such as Leigh syndrome, for which there are no effective therapies. We found that rapamycin, a specific inhibitor of the mechanistic target of rapamycin (mTOR) signaling pathway, robustly enhances survival and attenuates disease progression in a mouse model of Leigh syndrome. Administration of rapamycin to these mice, which are deficient in the mitochondrial respiratory chain subunit Ndufs4 [NADH dehydrogenase (ubiquinone) Fe-S protein 4], delays onset of neurological symptoms, reduces neuroinflammation, and prevents brain lesions. Although the precise mechanism of rescue remains to be determined, rapamycin induces a metabolic shift toward amino acid catabolism and away from glycolysis, alleviating the buildup of glycolytic intermediates. This therapeutic strategy may prove relevant for a broad range of mitochondrial diseases.
Figures
Comment in
-
Neurometabolic disease: Treating mitochondrial diseases with mTOR inhibitors--a potential treatment for Leigh syndrome?Nat Rev Neurol. 2014 Jan;10(1):2. doi: 10.1038/nrneurol.2013.251. Epub 2013 Dec 3. Nat Rev Neurol. 2014. PMID: 24296657 No abstract available.
-
Medicine. A common pathway for a rare disease?Science. 2013 Dec 20;342(6165):1453-4. doi: 10.1126/science.1248449. Science. 2013. PMID: 24357304 No abstract available.
Similar articles
-
Medicine. A common pathway for a rare disease?Science. 2013 Dec 20;342(6165):1453-4. doi: 10.1126/science.1248449. Science. 2013. PMID: 24357304 No abstract available.
-
Neurometabolic disease: Treating mitochondrial diseases with mTOR inhibitors--a potential treatment for Leigh syndrome?Nat Rev Neurol. 2014 Jan;10(1):2. doi: 10.1038/nrneurol.2013.251. Epub 2013 Dec 3. Nat Rev Neurol. 2014. PMID: 24296657 No abstract available.
-
PKC downregulation upon rapamycin treatment attenuates mitochondrial disease.Nat Metab. 2020 Dec;2(12):1472-1481. doi: 10.1038/s42255-020-00319-x. Epub 2020 Dec 14. Nat Metab. 2020. PMID: 33324011 Free PMC article.
-
Ndufs4 knockout mouse models of Leigh syndrome: pathophysiology and intervention.Brain. 2022 Mar 29;145(1):45-63. doi: 10.1093/brain/awab426. Brain. 2022. PMID: 34849584 Free PMC article. Review.
-
Infantile mitochondrial disorders.Biosci Rep. 2007 Jun;27(1-3):105-12. doi: 10.1007/s10540-007-9039-y. Biosci Rep. 2007. PMID: 17486440 Review.
Cited by
-
The mTOR signalling cascade: paving new roads to cure neurological disease.Nat Rev Neurol. 2016 Jul;12(7):379-92. doi: 10.1038/nrneurol.2016.81. Epub 2016 Jun 24. Nat Rev Neurol. 2016. PMID: 27340022 Review.
-
Rejuvenating immunity: "anti-aging drug today" eight years later.Oncotarget. 2015 Aug 14;6(23):19405-12. doi: 10.18632/oncotarget.3740. Oncotarget. 2015. PMID: 25844603 Free PMC article.
-
Development of iPSC-based clinical trial selection platform for patients with ultrarare diseases.Sci Adv. 2022 Apr 8;8(14):eabl4370. doi: 10.1126/sciadv.abl4370. Epub 2022 Apr 8. Sci Adv. 2022. PMID: 35394834 Free PMC article.
-
Amino acids and amino acid sensing: implication for aging and diseases.Biogerontology. 2019 Feb;20(1):17-31. doi: 10.1007/s10522-018-9770-8. Epub 2018 Sep 25. Biogerontology. 2019. PMID: 30255223 Review.
-
Comparative mitochondrial genomics reveals a possible role of a recent duplication of NADH dehydrogenase subunit 5 in gene regulation.DNA Res. 2018 Dec 1;25(6):577-586. doi: 10.1093/dnares/dsy026. DNA Res. 2018. PMID: 30085012 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
