

Role of endothelial to mesenchymal transition in the pathogenesis of the vascular alterations in systemic sclerosis
- PMID: 24175099
- PMCID: PMC3794556
- DOI: 10.1155/2013/835948
Role of endothelial to mesenchymal transition in the pathogenesis of the vascular alterations in systemic sclerosis
Abstract
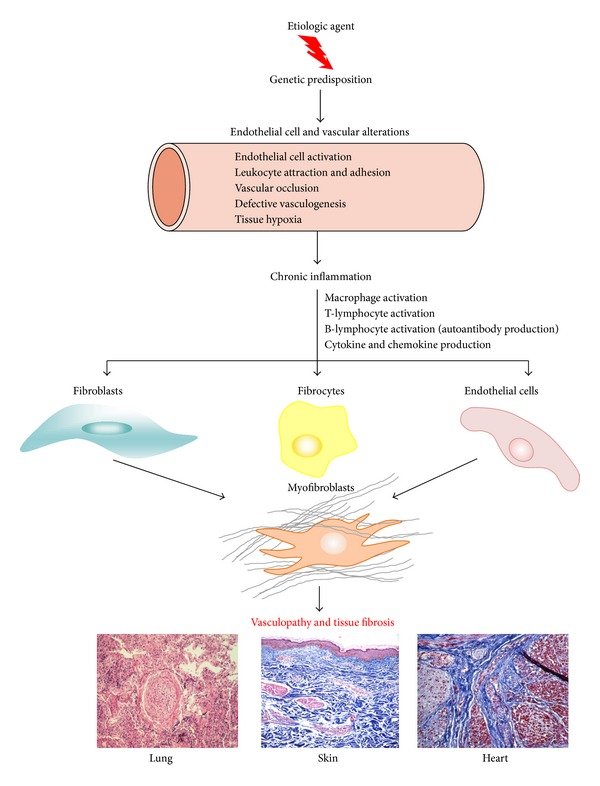
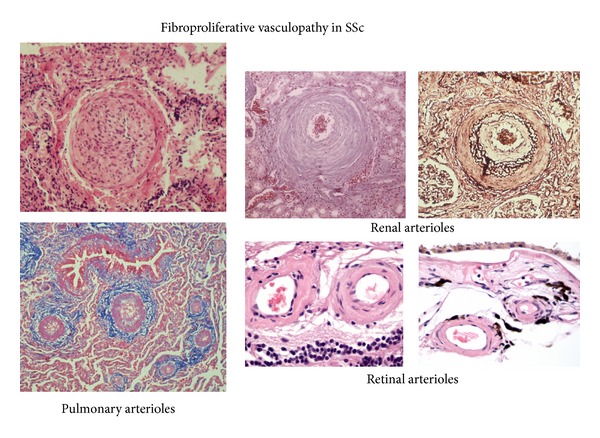
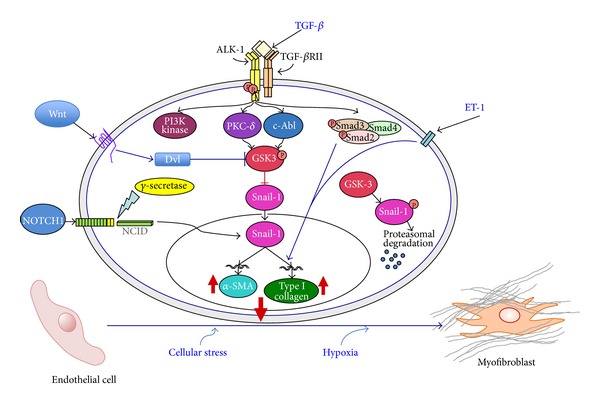
The pathogenesis of Systemic Sclerosis (SSc) is extremely complex, and despite extensive studies, the exact mechanisms involved are not well understood. Numerous recent studies of early events in SSc pathogenesis have suggested that unknown etiologic factors in a genetically receptive host trigger structural and functional microvascular endothelial cell abnormalities. These alterations result in the attraction, transmigration, and accumulation of immune and inflammatory cells in the perivascular tissues, which in turn induce the phenotypic conversion of endothelial cells and quiescent fibroblasts into activated myofibroblasts, a process known as endothelial to mesenchymal transition or EndoMT. The activated myofibroblasts are the effector cells responsible for the severe and frequently progressive fibrotic process and the fibroproliferative vasculopathy that are the hallmarks of SSc. Thus, according to this hypothesis the endothelial and vascular alterations, which include the phenotypic conversion of endothelial cells into activated myofibroblasts, play a crucial role in the development of the progressive fibrotic process affecting skin and multiple internal organs. The role of endothelial cell and vascular alterations, the potential contribution of endothelial to mesenchymal cell transition in the pathogenesis of the tissue fibrosis, and fibroproliferative vasculopathy in SSc will be reviewed here.
Figures
Similar articles
-
Endothelial to mesenchymal transition (EndoMT) in the pathogenesis of Systemic Sclerosis-associated pulmonary fibrosis and pulmonary arterial hypertension. Myth or reality?Matrix Biol. 2016 Apr;51:26-36. doi: 10.1016/j.matbio.2016.01.012. Epub 2016 Jan 22. Matrix Biol. 2016. PMID: 26807760 Free PMC article. Review.
-
Endothelial to Mesenchymal Transition (EndoMT) in the Pathogenesis of Human Fibrotic Diseases.J Clin Med. 2016 Apr 11;5(4):45. doi: 10.3390/jcm5040045. J Clin Med. 2016. PMID: 27077889 Free PMC article. Review.
-
New Insights into Profibrotic Myofibroblast Formation in Systemic Sclerosis: When the Vascular Wall Becomes the Enemy.Life (Basel). 2021 Jun 24;11(7):610. doi: 10.3390/life11070610. Life (Basel). 2021. PMID: 34202703 Free PMC article. Review.
-
Role of microRNA in the pathogenesis of systemic sclerosis tissue fibrosis and vasculopathy.Autoimmun Rev. 2019 Nov;18(11):102396. doi: 10.1016/j.autrev.2019.102396. Epub 2019 Sep 11. Autoimmun Rev. 2019. PMID: 31520794 Review.
-
Cellular Transdifferentiation: A Crucial Mechanism of Fibrosis in Systemic Sclerosis.Curr Rheumatol Rev. 2024;20(4):388-404. doi: 10.2174/0115733971261932231025045400. Curr Rheumatol Rev. 2024. PMID: 37921216 Review.
Cited by
-
A Role of Myocardin Related Transcription Factor-A (MRTF-A) in Scleroderma Related Fibrosis.PLoS One. 2015 May 8;10(5):e0126015. doi: 10.1371/journal.pone.0126015. eCollection 2015. PLoS One. 2015. PMID: 25955164 Free PMC article.
-
Endothelial-to-mesenchymal transition: An underappreciated mediator of diabetic complications.Front Endocrinol (Lausanne). 2023 Jan 27;14:1050540. doi: 10.3389/fendo.2023.1050540. eCollection 2023. Front Endocrinol (Lausanne). 2023. PMID: 36777351 Free PMC article. Review.
-
Endothelial dysfunction: the role of sterol regulatory element-binding protein-induced NOD-like receptor family pyrin domain-containing protein 3 inflammasome in atherosclerosis.Curr Opin Lipidol. 2014 Oct;25(5):339-49. doi: 10.1097/MOL.0000000000000107. Curr Opin Lipidol. 2014. PMID: 25188917 Free PMC article. Review.
-
Immune and Non-Immune Inflammatory Cells Involved in Autoimmune Fibrosis: New Discoveries.J Clin Med. 2023 May 31;12(11):3801. doi: 10.3390/jcm12113801. J Clin Med. 2023. PMID: 37297996 Free PMC article. Review.
-
Systems-based identification of the Hippo pathway for promoting fibrotic mesenchymal differentiation in systemic sclerosis.Nat Commun. 2024 Jan 3;15(1):210. doi: 10.1038/s41467-023-44645-6. Nat Commun. 2024. PMID: 38172207 Free PMC article.
References
-
- Gabrielli A, Avvedimento EV, Krieg T. Scleroderma. The New England Journal of Medicine. 2009;360(19):1989–2003. - PubMed
-
- Jimenez SA, Derk CT. Following the molecular pathways toward an understanding of the pathogenesis of systemic sclerosis. Annals of Internal Medicine. 2004;140(1):37–50. - PubMed
-
- Katsumoto TR, Whitfield ML, Connolly MK. The pathogenesis of systemic sclerosis. Annual Review of Pathology: Mechanisms of Disease. 2011;6:509–537. - PubMed
-
- Denton CP, Black CM, Abraham DJ. Mechanisms and consequences of fibrosis in systemic sclerosis. Nature Clinical Practice Rheumatology. 2006;2:134–144. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
