

Ubiquitously transcribed genes use alternative polyadenylation to achieve tissue-specific expression
- PMID: 24145798
- PMCID: PMC3828523
- DOI: 10.1101/gad.229328.113
Ubiquitously transcribed genes use alternative polyadenylation to achieve tissue-specific expression
Abstract
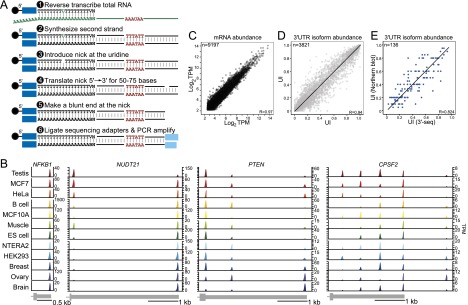
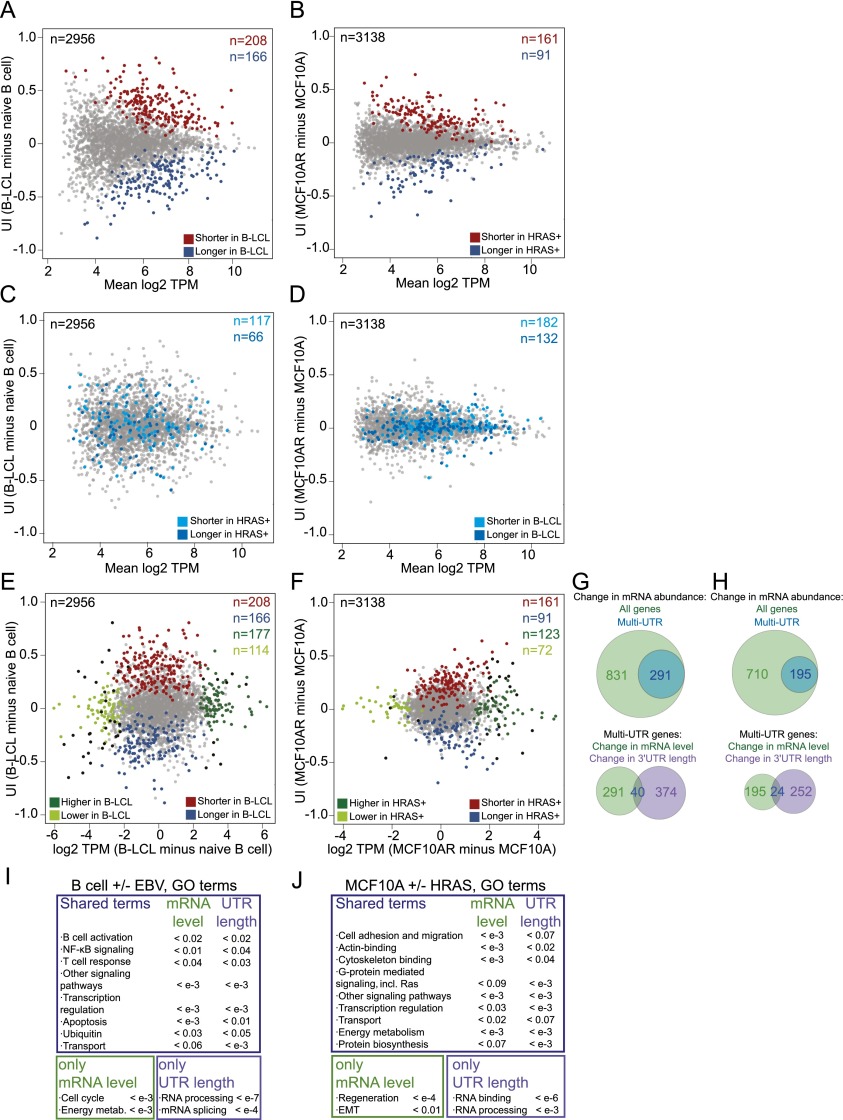
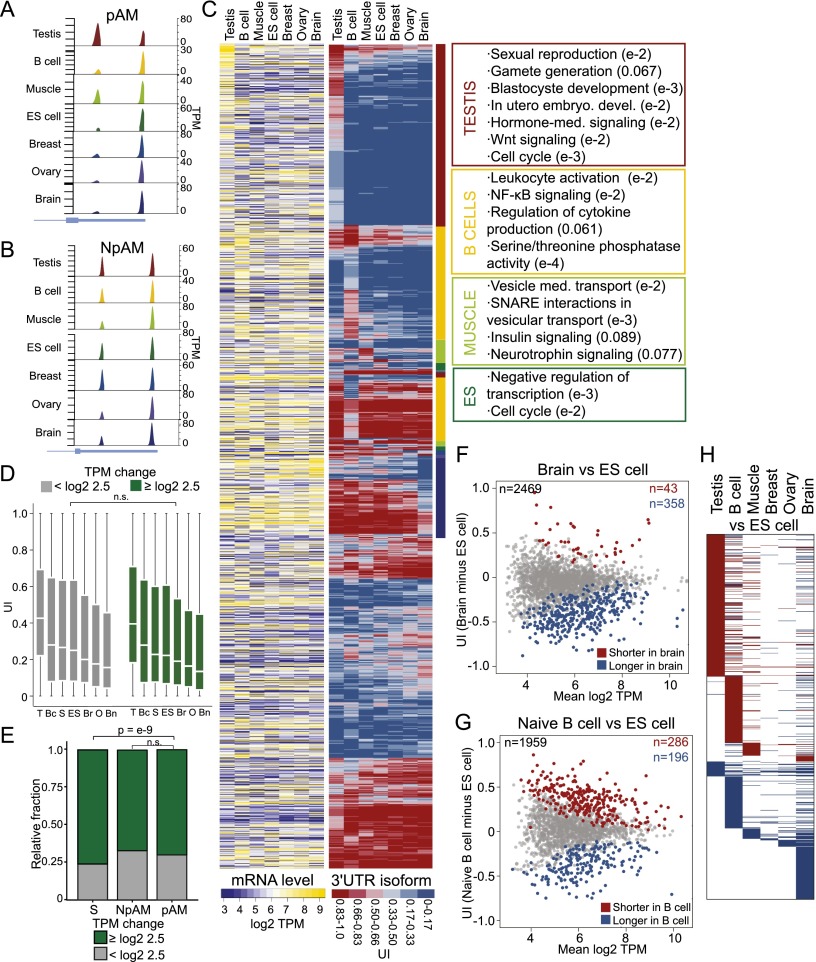
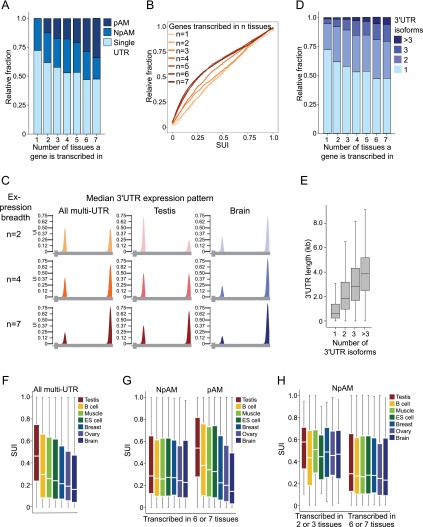
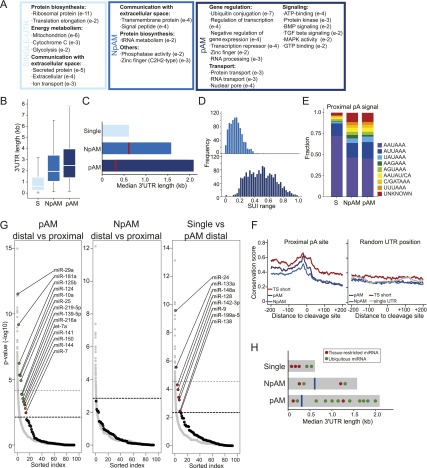
More than half of human genes use alternative cleavage and polyadenylation (ApA) to generate mRNA transcripts that differ in the lengths of their 3' untranslated regions (UTRs), thus altering the post-transcriptional fate of the message and likely the protein output. The extent of 3' UTR variation across tissues and the functional role of ApA remain poorly understood. We developed a sequencing method called 3'-seq to quantitatively map the 3' ends of the transcriptome of diverse human tissues and isogenic transformation systems. We found that cell type-specific gene expression is accomplished by two complementary programs. Tissue-restricted genes tend to have single 3' UTRs, whereas a majority of ubiquitously transcribed genes generate multiple 3' UTRs. During transformation and differentiation, single-UTR genes change their mRNA abundance levels, while multi-UTR genes mostly change 3' UTR isoform ratios to achieve tissue specificity. However, both regulation programs target genes that function in the same pathways and processes that characterize the new cell type. Instead of finding global shifts in 3' UTR length during transformation and differentiation, we identify tissue-specific groups of multi-UTR genes that change their 3' UTR ratios; these changes in 3' UTR length are largely independent from changes in mRNA abundance. Finally, tissue-specific usage of ApA sites appears to be a mechanism for changing the landscape targetable by ubiquitously expressed microRNAs.
Keywords: 3′ UTR isoform; alternative polyadenylation; computational biology; gene regulation; tissue-specific regulation of gene expression; transcriptome analysis.
Figures
Similar articles
-
APA-Scan: detection and visualization of 3'-UTR alternative polyadenylation with RNA-seq and 3'-end-seq data.BMC Bioinformatics. 2022 Sep 28;23(Suppl 3):396. doi: 10.1186/s12859-022-04939-w. BMC Bioinformatics. 2022. PMID: 36171568 Free PMC article.
-
Shortening of 3' UTRs in most cell types composing tumor tissues implicates alternative polyadenylation in protein metabolism.RNA. 2021 Dec;27(12):1459-1470. doi: 10.1261/rna.078886.121. Epub 2021 Sep 14. RNA. 2021. PMID: 34521731 Free PMC article.
-
Global estimation of the 3' untranslated region landscape using RNA sequencing.Methods. 2015 Jul 15;83:111-7. doi: 10.1016/j.ymeth.2015.04.011. Epub 2015 Apr 18. Methods. 2015. PMID: 25899044
-
Implications of polyadenylation in health and disease.Nucleus. 2014;5(6):508-19. doi: 10.4161/nucl.36360. Epub 2014 Oct 31. Nucleus. 2014. PMID: 25484187 Free PMC article. Review.
-
Alternative Polyadenylation in Stem Cell Self-Renewal and Differentiation.Trends Mol Med. 2021 Jul;27(7):660-672. doi: 10.1016/j.molmed.2021.04.006. Epub 2021 May 11. Trends Mol Med. 2021. PMID: 33985920 Review.
Cited by
-
Genome-wide dynamics of alternative polyadenylation in rice.Genome Res. 2016 Dec;26(12):1753-1760. doi: 10.1101/gr.210757.116. Epub 2016 Oct 12. Genome Res. 2016. PMID: 27733415 Free PMC article.
-
Homo sapiens exhibit a distinct pattern of CNV genes regulation: an important role of miRNAs and SNPs in expression plasticity.Sci Rep. 2015 Jul 16;5:12163. doi: 10.1038/srep12163. Sci Rep. 2015. PMID: 26178010 Free PMC article.
-
Altered RNA Processing in Cancer Pathogenesis and Therapy.Cancer Discov. 2019 Nov;9(11):1493-1510. doi: 10.1158/2159-8290.CD-19-0399. Epub 2019 Oct 14. Cancer Discov. 2019. PMID: 31611195 Free PMC article. Review.
-
Phase-Separated Subcellular Compartmentation and Related Human Diseases.Int J Mol Sci. 2022 May 14;23(10):5491. doi: 10.3390/ijms23105491. Int J Mol Sci. 2022. PMID: 35628304 Free PMC article. Review.
-
CRISPR-Mediated Knockout of Long 3' UTR mRNA Isoforms in mESC-Derived Neurons.Front Genet. 2021 Dec 17;12:789434. doi: 10.3389/fgene.2021.789434. eCollection 2021. Front Genet. 2021. PMID: 34976020 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources