

Opposing action of nuclear factor κB and Polo-like kinases determines a homeostatic end point for excitatory synaptic adaptation
- PMID: 24133254
- PMCID: PMC3797372
- DOI: 10.1523/JNEUROSCI.2131-13.2013
Opposing action of nuclear factor κB and Polo-like kinases determines a homeostatic end point for excitatory synaptic adaptation
Abstract
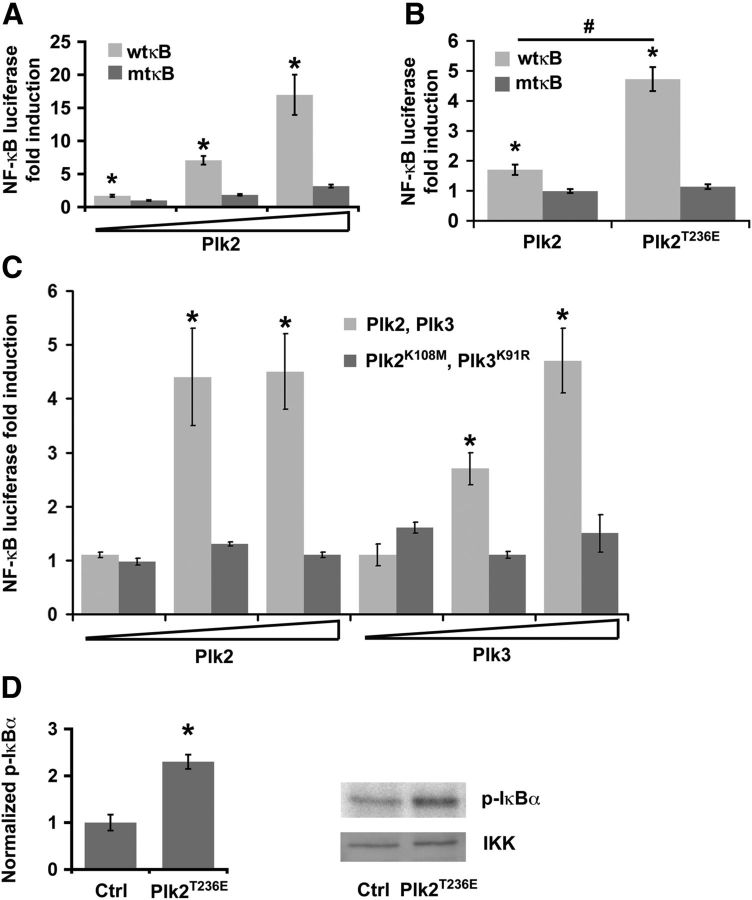
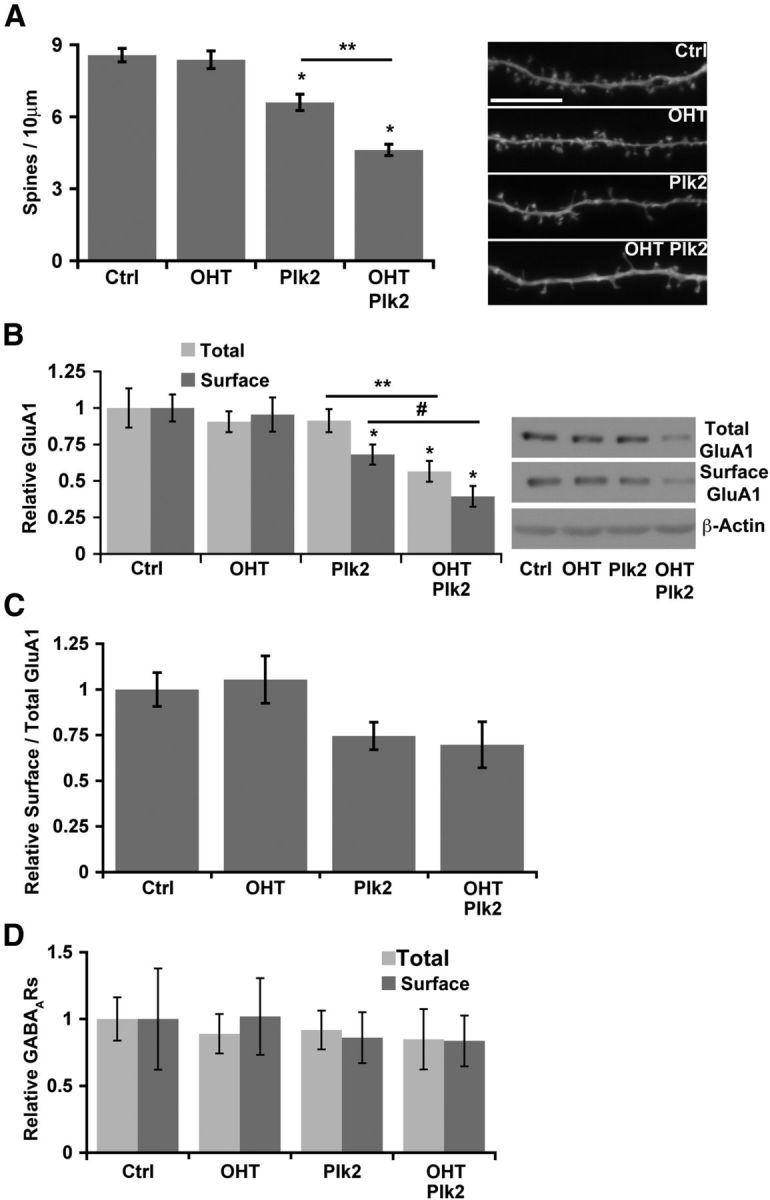
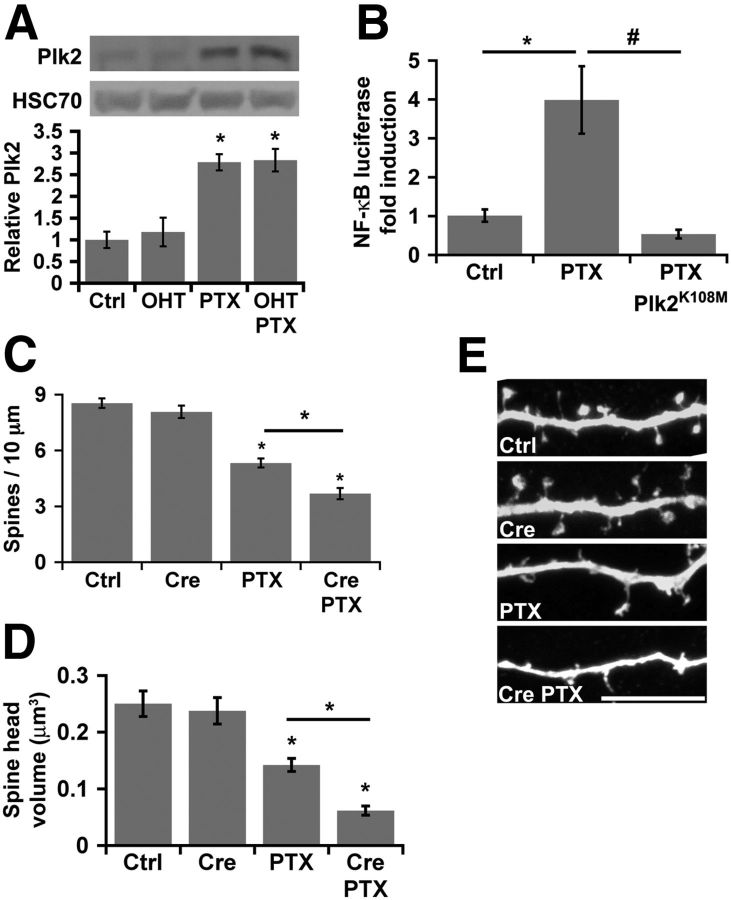
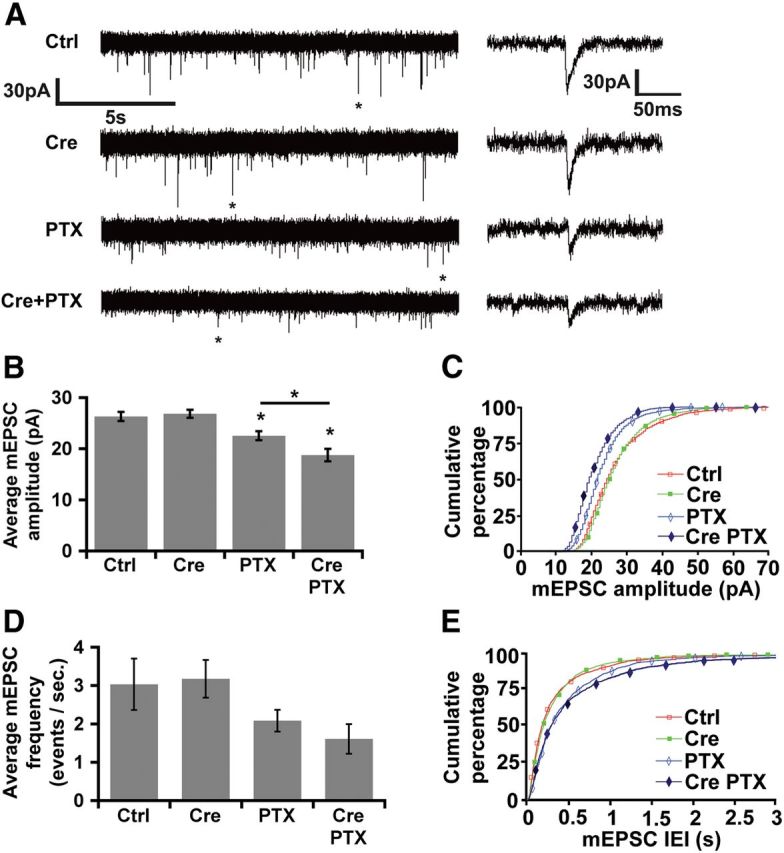
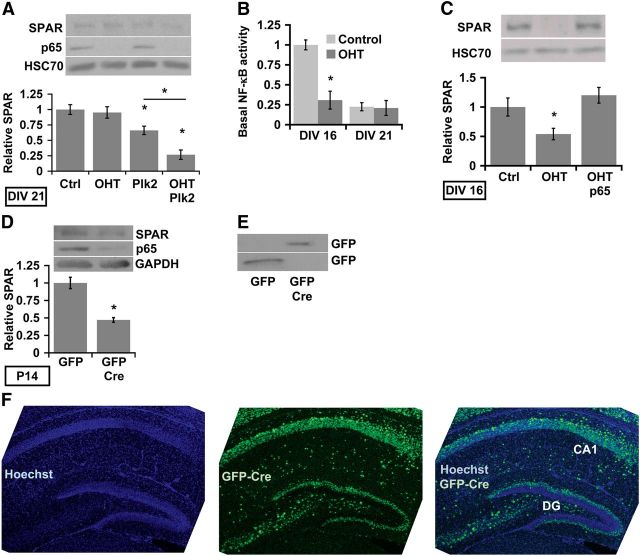
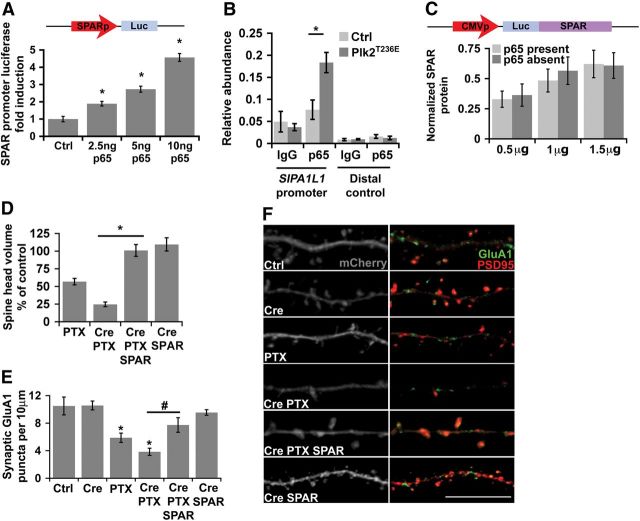
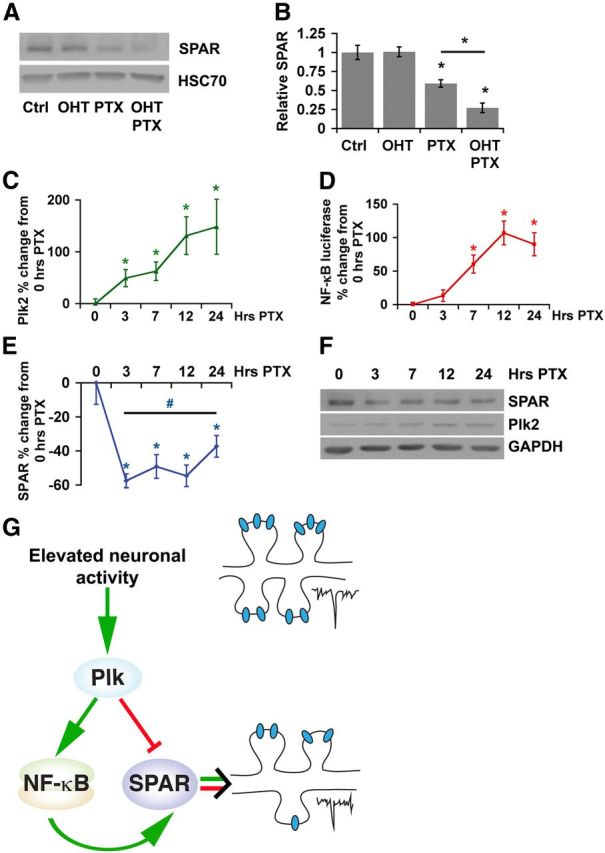
Homeostatic responses critically adjust synaptic strengths to maintain stability in neuronal networks. Compensatory adaptations to prolonged excitation include induction of Polo-like kinases (Plks) and degradation of spine-associated Rap GTPase-activating protein (SPAR) to reduce synaptic excitation, but mechanisms that limit overshooting and allow refinement of homeostatic adjustments remain poorly understood. We report that Plks produce canonical pathway-mediated activation of the nuclear factor κB (NF-κB) transcription factor in a process that requires the kinase activity of Plks. Chronic elevated activity, which induces Plk expression, also produces Plk-dependent activation of NF-κB. Deficiency of NF-κB, in the context of exogenous Plk2 expression or chronic elevated neuronal excitation, produces exaggerated homeostatic reductions in the size and density of dendritic spines, synaptic AMPA glutamate receptor levels, and excitatory synaptic currents. During the homeostatic response to chronic elevated activity, NF-κB activation by Plks subsequently opposes Plk-mediated SPAR degradation by transcriptionally upregulating SPAR in mouse hippocampal neurons in vitro and in vivo. Exogenous SPAR expression can rescue the overshooting of homeostatic reductions at excitatory synapses in NF-κB-deficient neurons responding to elevated activity. Our data establish an integral feedback loop involving NF-κB, Plks, and SPAR that regulates the end point of homeostatic synaptic adaptation to elevated activity and are the first to implicate a transcription factor in the regulation of homeostatic synaptic responses.
Figures
Similar articles
-
IκB kinase/nuclear factor κB-dependent insulin-like growth factor 2 (Igf2) expression regulates synapse formation and spine maturation via Igf2 receptor signaling.J Neurosci. 2012 Apr 18;32(16):5688-703. doi: 10.1523/JNEUROSCI.0111-12.2012. J Neurosci. 2012. PMID: 22514330 Free PMC article.
-
Polo-like kinases in the nervous system.Oncogene. 2005 Jan 10;24(2):292-8. doi: 10.1038/sj.onc.1208277. Oncogene. 2005. PMID: 15640845 Review.
-
Targeting of NF-κB to Dendritic Spines Is Required for Synaptic Signaling and Spine Development.J Neurosci. 2018 Apr 25;38(17):4093-4103. doi: 10.1523/JNEUROSCI.2663-16.2018. Epub 2018 Mar 19. J Neurosci. 2018. PMID: 29555853 Free PMC article.
-
A requirement for nuclear factor-kappaB in developmental and plasticity-associated synaptogenesis.J Neurosci. 2011 Apr 6;31(14):5414-25. doi: 10.1523/JNEUROSCI.2456-10.2011. J Neurosci. 2011. PMID: 21471377 Free PMC article.
-
Regulation of neuronal PKA signaling through AKAP targeting dynamics.Eur J Cell Biol. 2006 Jul;85(7):627-33. doi: 10.1016/j.ejcb.2006.01.010. Epub 2006 Feb 28. Eur J Cell Biol. 2006. PMID: 16504338 Review.
Cited by
-
Neuronal Gene Targets of NF-κB and Their Dysregulation in Alzheimer's Disease.Front Mol Neurosci. 2016 Nov 9;9:118. doi: 10.3389/fnmol.2016.00118. eCollection 2016. Front Mol Neurosci. 2016. PMID: 27881951 Free PMC article. Review.
-
Cellular Specificity of NF-κB Function in the Nervous System.Front Immunol. 2019 May 9;10:1043. doi: 10.3389/fimmu.2019.01043. eCollection 2019. Front Immunol. 2019. PMID: 31143184 Free PMC article. Review.
-
Imbalance between firing homeostasis and synaptic plasticity drives early-phase Alzheimer's disease.Nat Neurosci. 2018 Apr;21(4):463-473. doi: 10.1038/s41593-018-0080-x. Epub 2018 Feb 5. Nat Neurosci. 2018. PMID: 29403035 Free PMC article. Review.
-
Pharmacological inhibition of PLK2 kinase activity mitigates cognitive decline but aggravates APP pathology in a sex-dependent manner in APP/PS1 mouse model of Alzheimer's disease.Heliyon. 2024 Oct 18;10(20):e39571. doi: 10.1016/j.heliyon.2024.e39571. eCollection 2024 Oct 30. Heliyon. 2024. PMID: 39498012 Free PMC article.
-
NFκB-inducing kinase inhibits NFκB activity specifically in neurons of the CNS.J Neurochem. 2016 Apr;137(2):154-63. doi: 10.1111/jnc.13526. Epub 2016 Mar 15. J Neurochem. 2016. PMID: 26778773 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous