

Function and evolution of DNA methylation in Nasonia vitripennis
- PMID: 24130511
- PMCID: PMC3794928
- DOI: 10.1371/journal.pgen.1003872
Function and evolution of DNA methylation in Nasonia vitripennis
Abstract
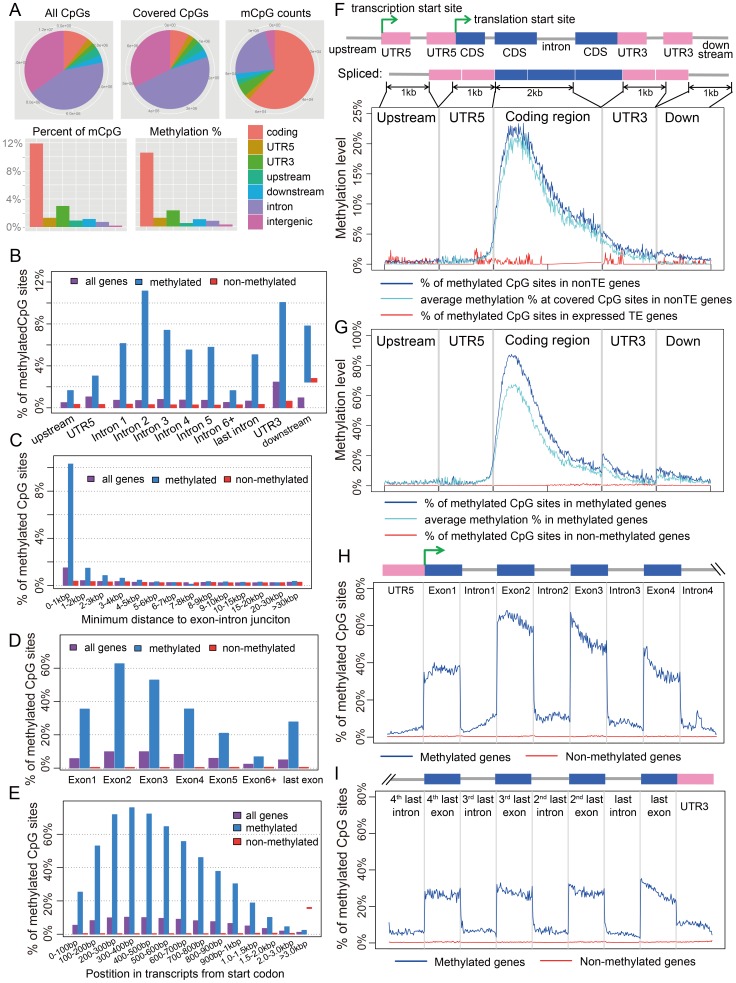
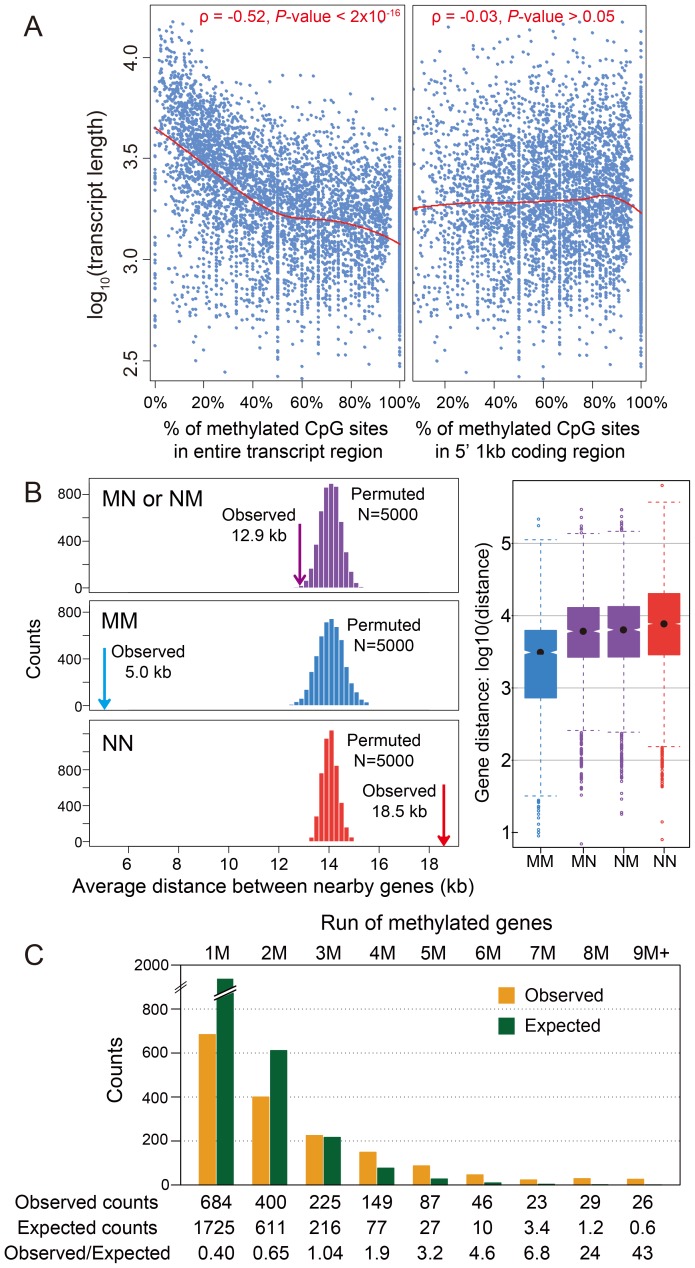
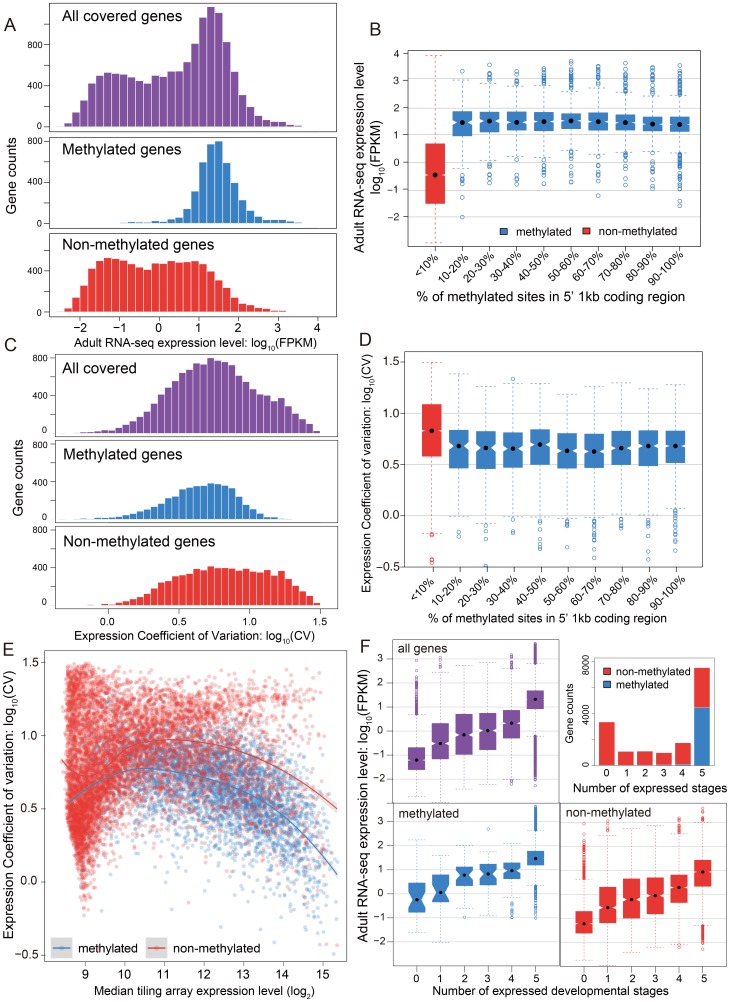
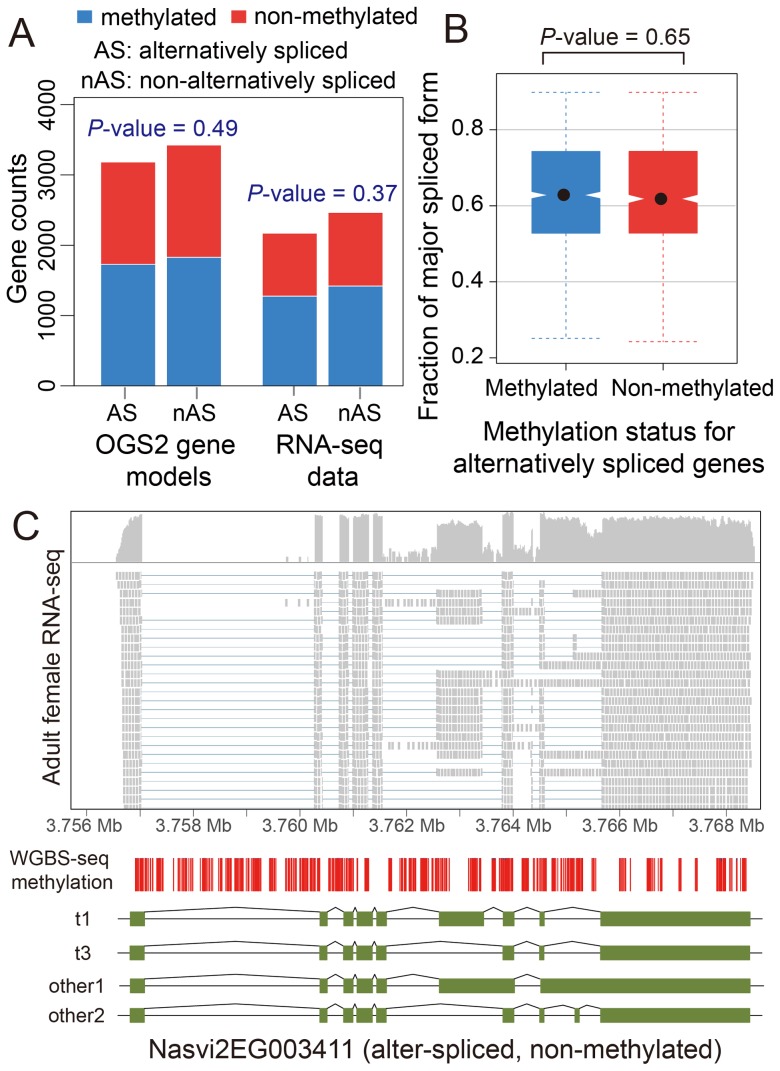
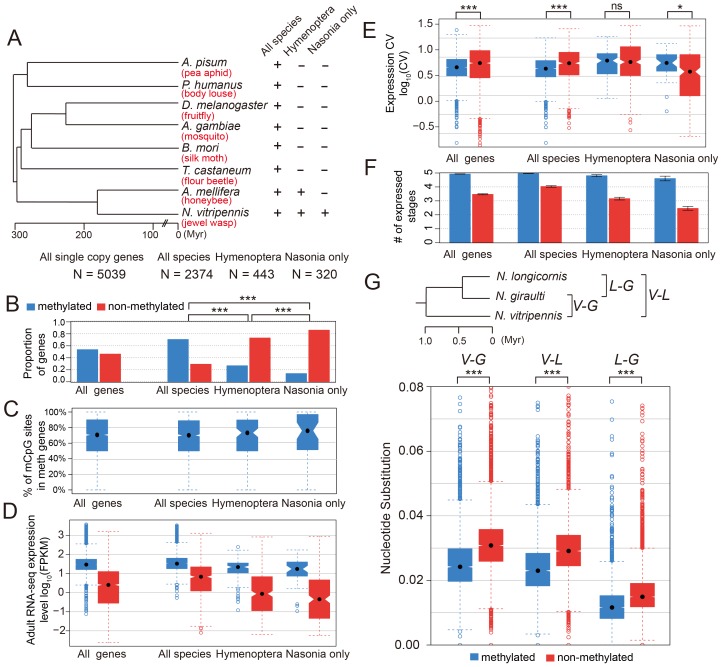
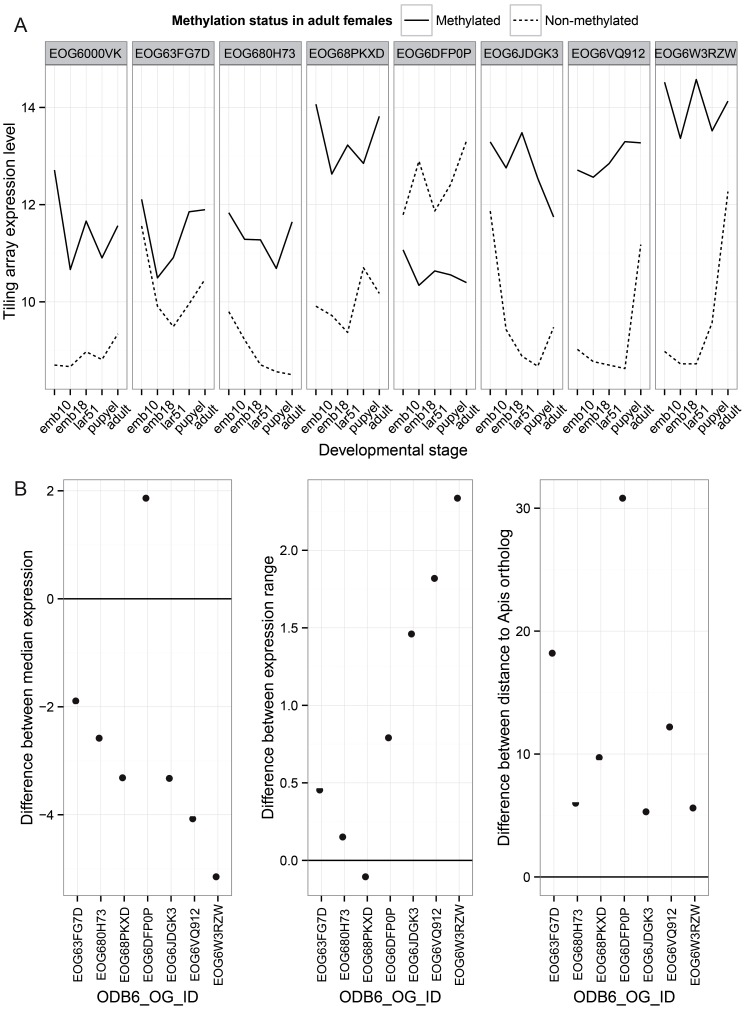
The parasitoid wasp Nasonia vitripennis is an emerging genetic model for functional analysis of DNA methylation. Here, we characterize genome-wide methylation at a base-pair resolution, and compare these results to gene expression across five developmental stages and to methylation patterns reported in other insects. An accurate assessment of DNA methylation across the genome is accomplished using bisulfite sequencing of adult females from a highly inbred line. One-third of genes show extensive methylation over the gene body, yet methylated DNA is not found in non-coding regions and rarely in transposons. Methylated genes occur in small clusters across the genome. Methylation demarcates exon-intron boundaries, with elevated levels over exons, primarily in the 5' regions of genes. It is also elevated near the sites of translational initiation and termination, with reduced levels in 5' and 3' UTRs. Methylated genes have higher median expression levels and lower expression variation across development stages than non-methylated genes. There is no difference in frequency of differential splicing between methylated and non-methylated genes, and as yet no established role for methylation in regulating alternative splicing in Nasonia. Phylogenetic comparisons indicate that many genes maintain methylation status across long evolutionary time scales. Nasonia methylated genes are more likely to be conserved in insects, but even those that are not conserved show broader expression across development than comparable non-methylated genes. Finally, examination of duplicated genes shows that those paralogs that have lost methylation in the Nasonia lineage following gene duplication evolve more rapidly, show decreased median expression levels, and increased specialization in expression across development. Methylation of Nasonia genes signals constitutive transcription across developmental stages, whereas non-methylated genes show more dynamic developmental expression patterns. We speculate that loss of methylation may result in increased developmental specialization in evolution and acquisition of methylation may lead to broader constitutive expression.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
Genome-wide association between DNA methylation and alternative splicing in an invertebrate.BMC Genomics. 2012 Sep 15;13:480. doi: 10.1186/1471-2164-13-480. BMC Genomics. 2012. PMID: 22978521 Free PMC article.
-
Comparative analyses of DNA methylation and sequence evolution using Nasonia genomes.Mol Biol Evol. 2011 Dec;28(12):3345-54. doi: 10.1093/molbev/msr168. Epub 2011 Jun 20. Mol Biol Evol. 2011. PMID: 21693438 Free PMC article.
-
Whole-genome DNA methylation profile of the jewel wasp (Nasonia vitripennis).G3 (Bethesda). 2014 Mar 20;4(3):383-8. doi: 10.1534/g3.113.008953. G3 (Bethesda). 2014. PMID: 24381191 Free PMC article.
-
Origin and evolution of spliceosomal introns.Biol Direct. 2012 Apr 16;7:11. doi: 10.1186/1745-6150-7-11. Biol Direct. 2012. PMID: 22507701 Free PMC article. Review.
-
DNA Methylation and Gene Regulation in Honeybees: From Genome-Wide Analyses to Obligatory Epialleles.Adv Exp Med Biol. 2016;945:193-211. doi: 10.1007/978-3-319-43624-1_9. Adv Exp Med Biol. 2016. PMID: 27826840 Review.
Cited by
-
Universality of the DNA methylation codes in Eucaryotes.Sci Rep. 2019 Jan 17;9(1):173. doi: 10.1038/s41598-018-37407-8. Sci Rep. 2019. PMID: 30655579 Free PMC article.
-
Genomic Landscape of Methylation Islands in Hymenopteran Insects.Genome Biol Evol. 2018 Oct 1;10(10):2766-2776. doi: 10.1093/gbe/evy203. Genome Biol Evol. 2018. PMID: 30239702 Free PMC article.
-
Male-Biased microRNA Discovery in the Pea Aphid.Insects. 2021 Jun 8;12(6):533. doi: 10.3390/insects12060533. Insects. 2021. PMID: 34201015 Free PMC article.
-
Modifications to gene body methylation do not alter gene expression plasticity in a reef-building coral.Evol Appl. 2024 Feb 22;17(2):e13662. doi: 10.1111/eva.13662. eCollection 2024 Feb. Evol Appl. 2024. PMID: 38390378 Free PMC article.
-
------Widespread conservation and lineage-specific diversification of genome-wide DNA methylation patterns across arthropods.PLoS Genet. 2020 Jun 25;16(6):e1008864. doi: 10.1371/journal.pgen.1008864. eCollection 2020 Jun. PLoS Genet. 2020. PMID: 32584820 Free PMC article.
References
-
- Jones PA (2012) Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet 13: 484–492. - PubMed
-
- Klose RJ, Bird AP (2006) Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci 31: 89–97. - PubMed
-
- Suzuki MM, Bird A (2008) DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet 9: 465–476. - PubMed
-
- Bird A (2002) DNA methylation patterns and epigenetic memory. Genes Dev 16: 6–21. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
