

Covariation between homeodomain transcription factors and the shape of their DNA binding sites
- PMID: 24078250
- PMCID: PMC3874178
- DOI: 10.1093/nar/gkt862
Covariation between homeodomain transcription factors and the shape of their DNA binding sites
Abstract
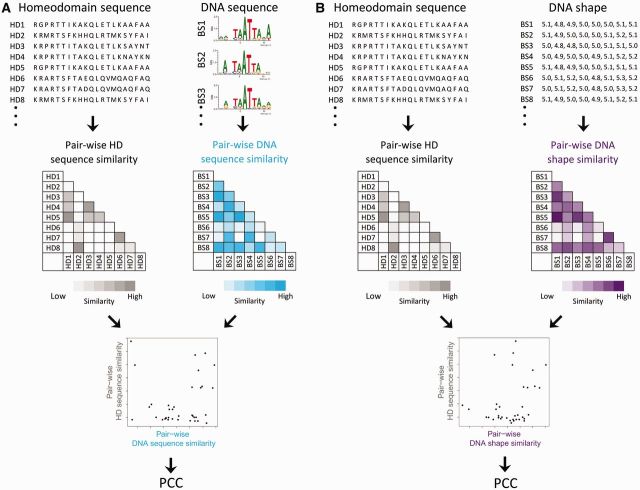
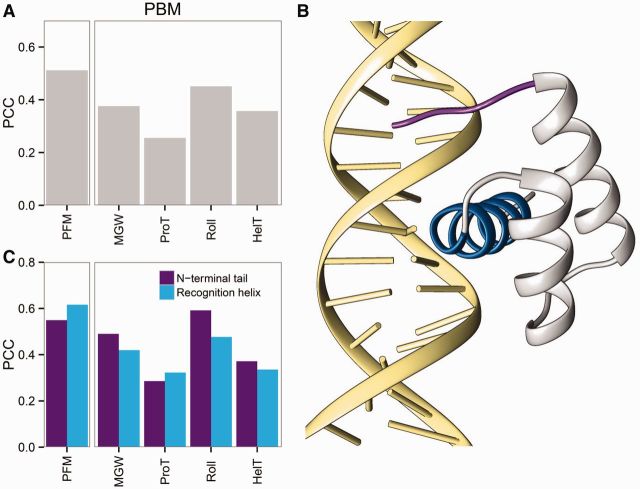
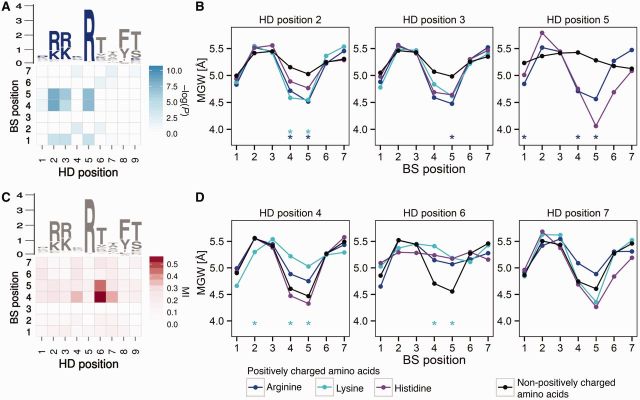
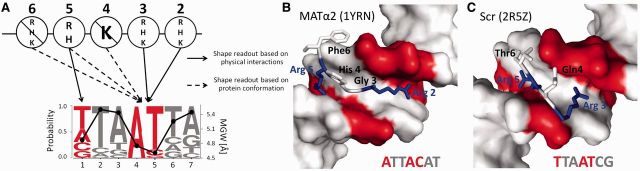
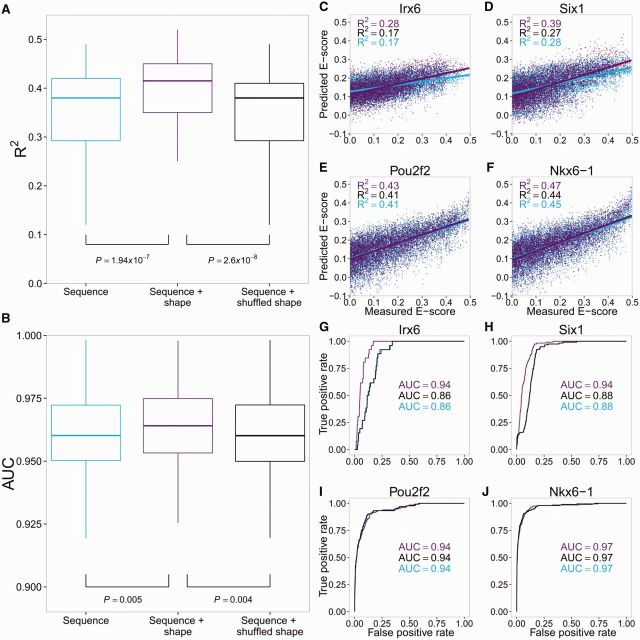
Protein-DNA recognition is a critical component of gene regulatory processes but the underlying molecular mechanisms are not yet completely understood. Whereas the DNA binding preferences of transcription factors (TFs) are commonly described using nucleotide sequences, the 3D DNA structure is recognized by proteins and is crucial for achieving binding specificity. However, the ability to analyze DNA shape in a high-throughput manner made it only recently feasible to integrate structural information into studies of protein-DNA binding. Here we focused on the homeodomain family of TFs and analyzed the DNA shape of thousands of their DNA binding sites, investigating the covariation between the protein sequence and the sequence and shape of their DNA targets. We found distinct homeodomain regions that were more correlated with either the nucleotide sequence or the DNA shape of their preferred binding sites, demonstrating different readout mechanisms through which homeodomains attain DNA binding specificity. We identified specific homeodomain residues that likely play key roles in DNA recognition via shape readout. Finally, we showed that adding DNA shape information when characterizing binding sites improved the prediction accuracy of homeodomain binding specificities. Taken together, our findings indicate that DNA shape information can generally provide new mechanistic insights into TF binding.
Figures
Similar articles
-
Context-Dependent Gene Regulation by Homeodomain Transcription Factor Complexes Revealed by Shape-Readout Deficient Proteins.Mol Cell. 2020 Apr 2;78(1):152-167.e11. doi: 10.1016/j.molcel.2020.01.027. Epub 2020 Feb 12. Mol Cell. 2020. PMID: 32053778 Free PMC article.
-
Deconvolving the recognition of DNA shape from sequence.Cell. 2015 Apr 9;161(2):307-18. doi: 10.1016/j.cell.2015.02.008. Epub 2015 Apr 2. Cell. 2015. PMID: 25843630 Free PMC article.
-
Interchange of DNA-binding modes in the deformed and ultrabithorax homeodomains: a structural role for the N-terminal arm.J Mol Biol. 2002 Nov 1;323(4):665-83. doi: 10.1016/s0022-2836(02)00996-8. J Mol Biol. 2002. PMID: 12419257
-
Homeodomain interactions.Curr Opin Struct Biol. 1996 Feb;6(1):62-8. doi: 10.1016/s0959-440x(96)80096-0. Curr Opin Struct Biol. 1996. PMID: 8696974 Review.
-
Transcription factors: the right combination for the DNA lock.Curr Biol. 1999 Jun 17;9(12):R456-8. doi: 10.1016/s0960-9822(99)80281-4. Curr Biol. 1999. PMID: 10375516 Review.
Cited by
-
Flexibility of flanking DNA is a key determinant of transcription factor affinity for the core motif.Biophys J. 2022 Oct 18;121(20):3987-4000. doi: 10.1016/j.bpj.2022.08.015. Epub 2022 Aug 17. Biophys J. 2022. PMID: 35978548 Free PMC article.
-
Dissecting the sharp response of a canonical developmental enhancer reveals multiple sources of cooperativity.Elife. 2019 Jun 21;8:e41266. doi: 10.7554/eLife.41266. Elife. 2019. PMID: 31223115 Free PMC article.
-
Geometric deep learning of protein-DNA binding specificity.Nat Methods. 2024 Sep;21(9):1674-1683. doi: 10.1038/s41592-024-02372-w. Epub 2024 Aug 5. Nat Methods. 2024. PMID: 39103447 Free PMC article.
-
Local sequence features that influence AP-1 cis-regulatory activity.Genome Res. 2018 Feb;28(2):171-181. doi: 10.1101/gr.226530.117. Epub 2018 Jan 5. Genome Res. 2018. PMID: 29305491 Free PMC article.
-
Predicting conformational ensembles and genome-wide transcription factor binding sites from DNA sequences.Sci Rep. 2017 Jun 22;7(1):4071. doi: 10.1038/s41598-017-03199-6. Sci Rep. 2017. PMID: 28642456 Free PMC article.
References
-
- Harris RC, Mackoy T, Dantas Machado AC, Xu D, Rohs R, Fenley MO. Chapter 3, vol. II In: T. Schlick (ed). Innovations in Biomolecular Modeling and Simulations. 2012. Opposites attract: shape and electrostatic complementarity in protein-DNA complexes. 53–80. The Royal Society of Chemistry. Cambridge, UK.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
