

Interleukin-1 and tumor necrosis factor-α trigger restriction of hepatitis B virus infection via a cytidine deaminase activation-induced cytidine deaminase (AID)
- PMID: 24025329
- PMCID: PMC3814766
- DOI: 10.1074/jbc.M113.501122
Interleukin-1 and tumor necrosis factor-α trigger restriction of hepatitis B virus infection via a cytidine deaminase activation-induced cytidine deaminase (AID)
Abstract
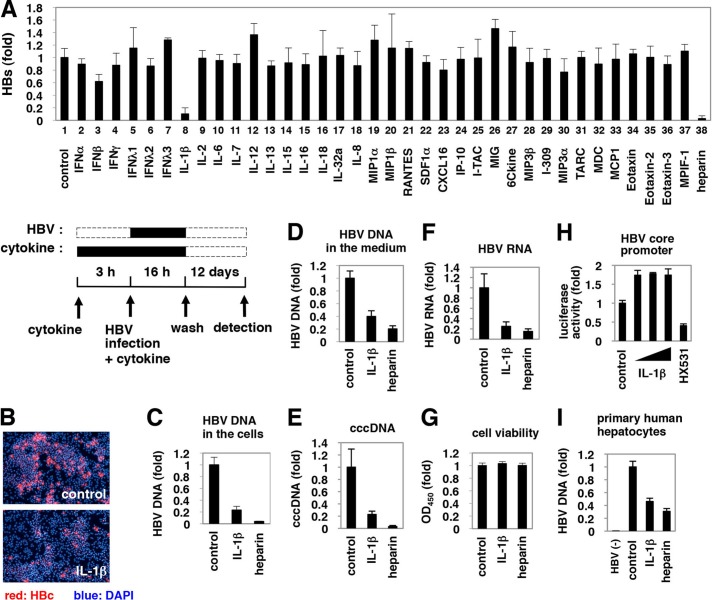
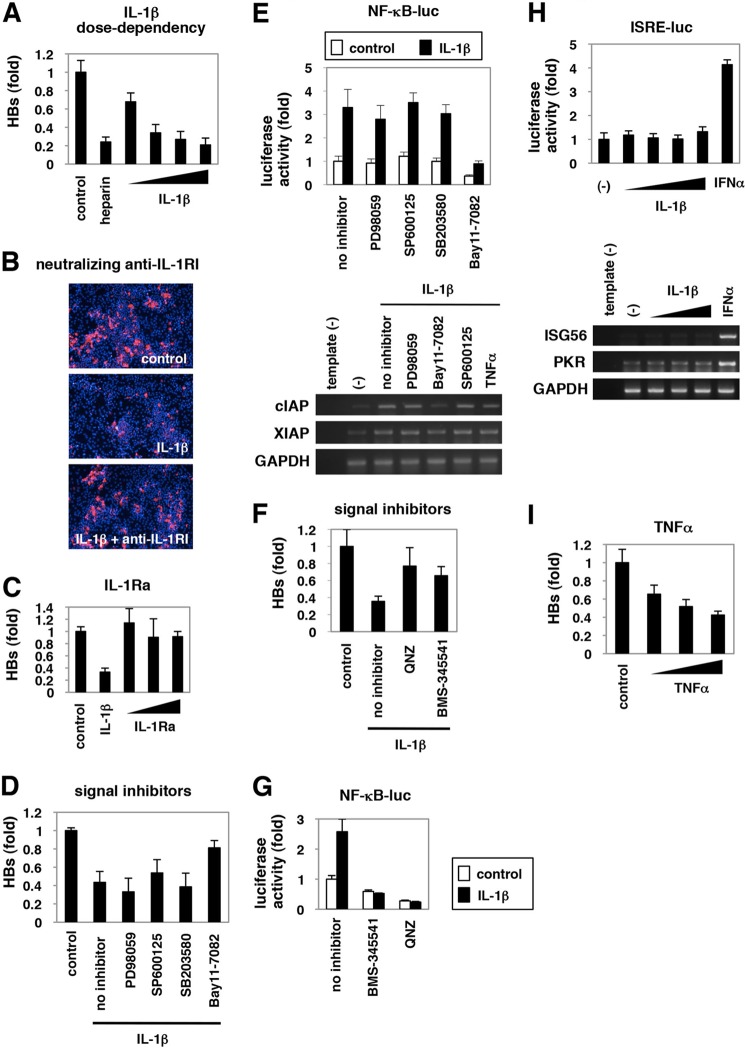
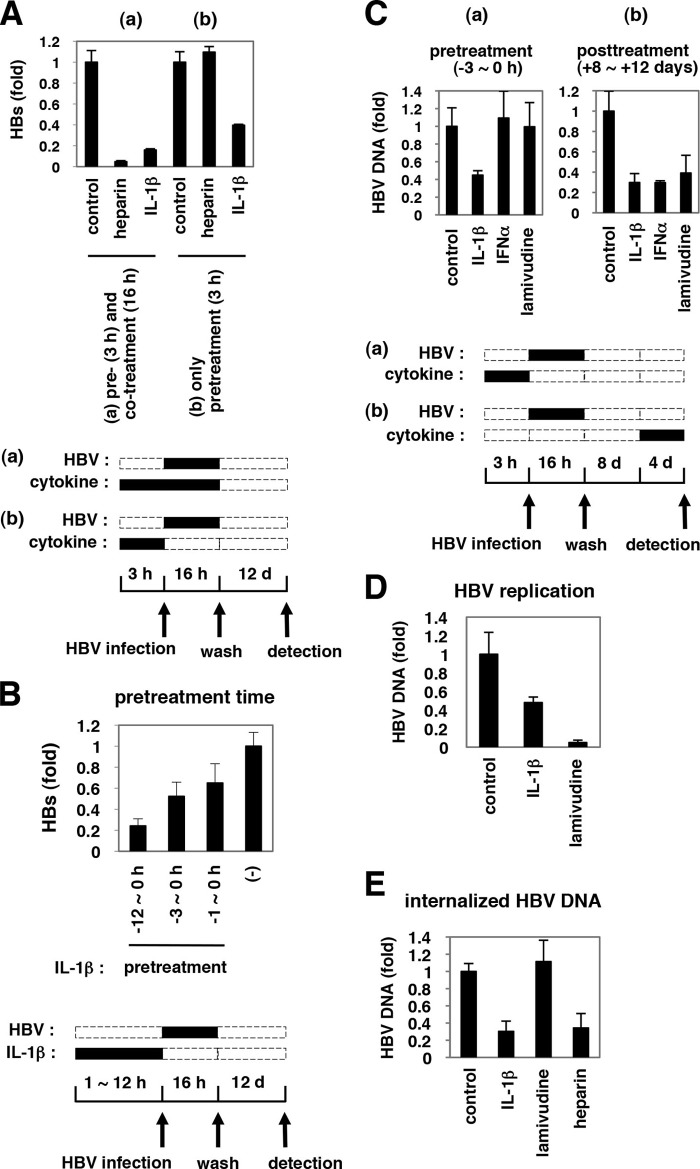
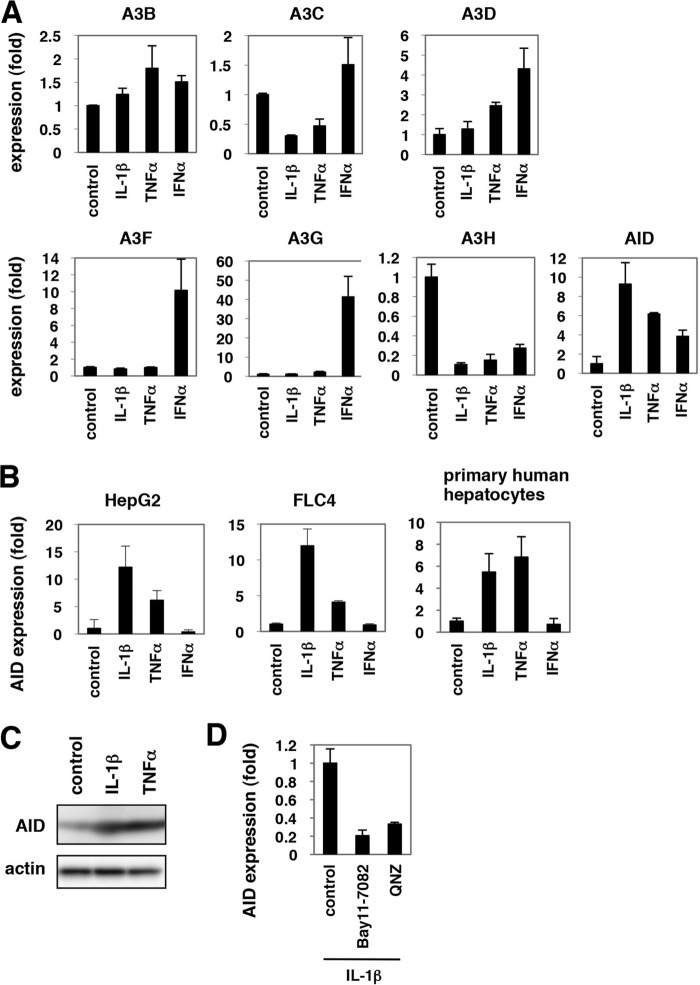
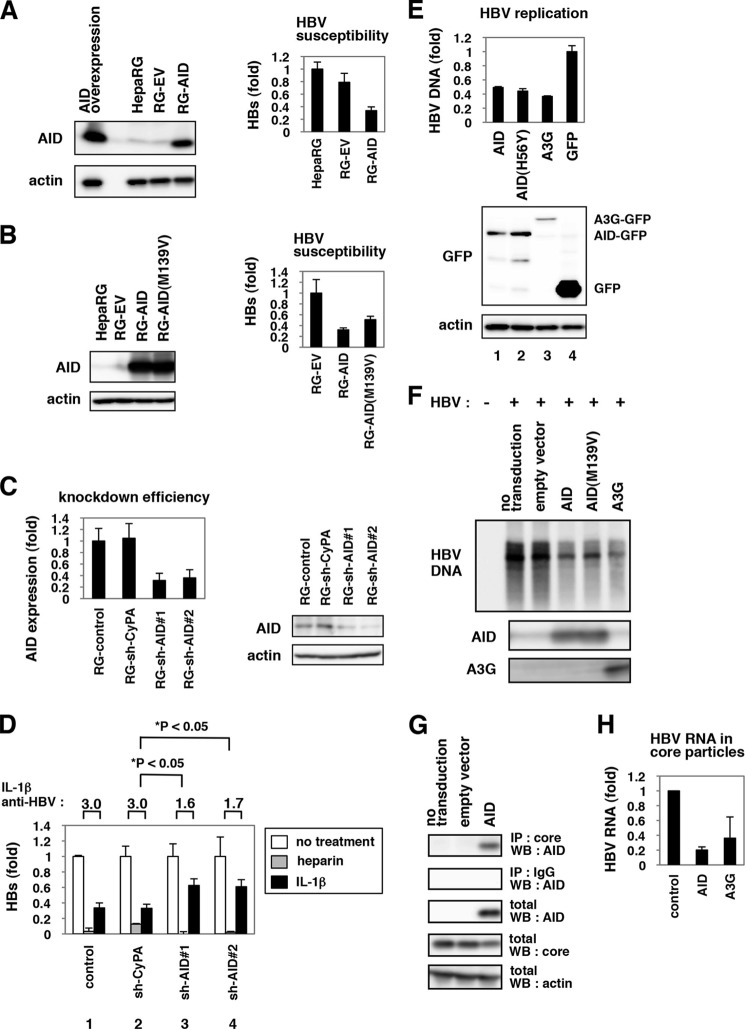
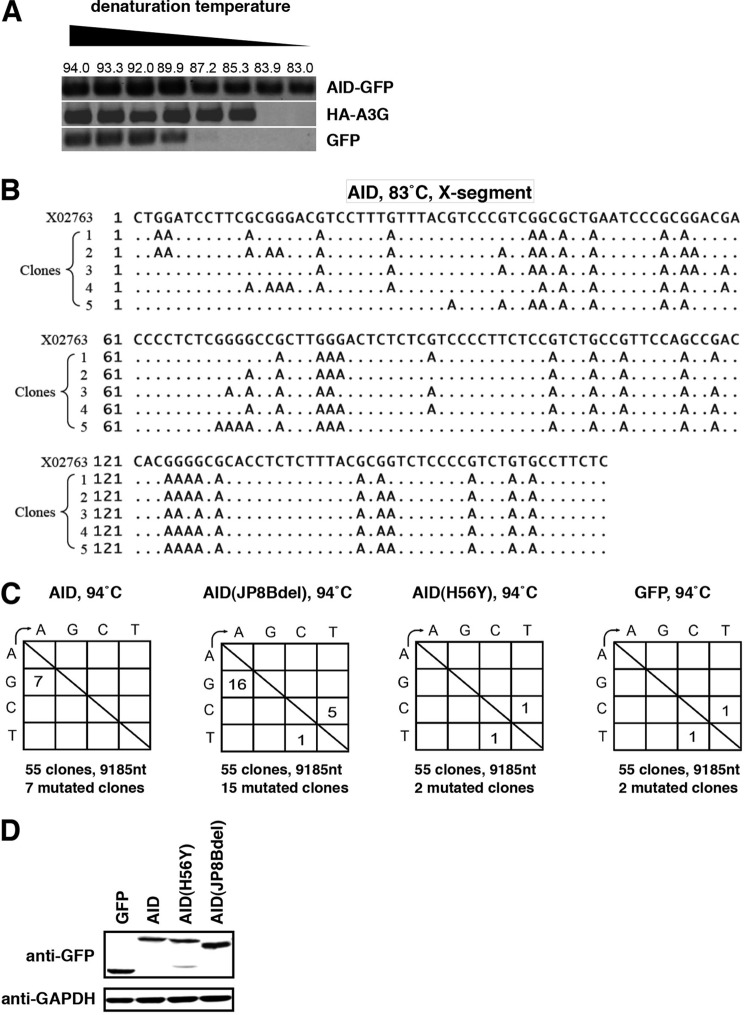
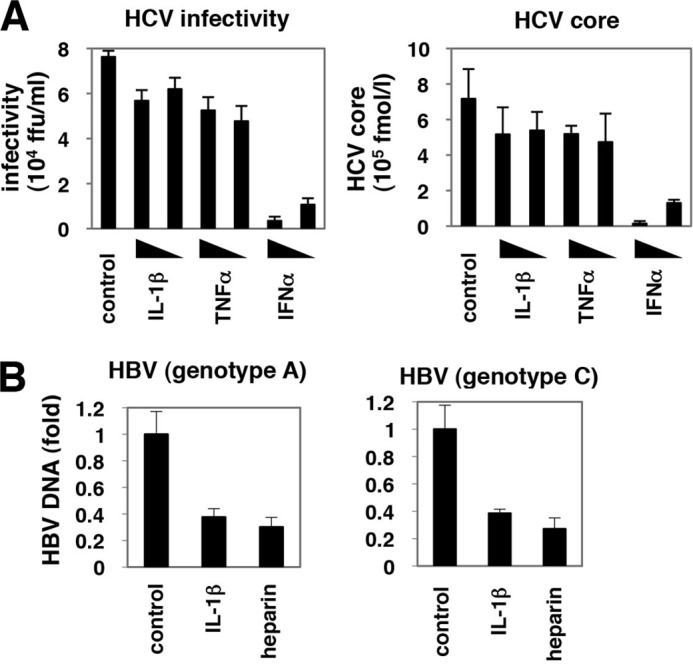
Virus infection is restricted by intracellular immune responses in host cells, and this is typically modulated by stimulation of cytokines. The cytokines and host factors that determine the host cell restriction against hepatitis B virus (HBV) infection are not well understood. We screened 36 cytokines and chemokines to determine which were able to reduce the susceptibility of HepaRG cells to HBV infection. Here, we found that pretreatment with IL-1β and TNFα remarkably reduced the host cell susceptibility to HBV infection. This effect was mediated by activation of the NF-κB signaling pathway. A cytidine deaminase, activation-induced cytidine deaminase (AID), was up-regulated by both IL-1β and TNFα in a variety of hepatocyte cell lines and primary human hepatocytes. Another deaminase APOBEC3G was not induced by these proinflammatory cytokines. Knockdown of AID expression impaired the anti-HBV effect of IL-1β, and overexpression of AID antagonized HBV infection, suggesting that AID was one of the responsible factors for the anti-HBV activity of IL-1/TNFα. Although AID induced hypermutation of HBV DNA, this activity was dispensable for the anti-HBV activity. The antiviral effect of IL-1/TNFα was also observed on different HBV genotypes but not on hepatitis C virus. These results demonstrate that proinflammatory cytokines IL-1/TNFα trigger a novel antiviral mechanism involving AID to regulate host cell permissiveness to HBV infection.
Keywords: AID; APOBEC3G; Deaminase; HBV; HepaRG; Innate Immunity; Interferon; Interleukin; Tumor Necrosis Factor (TNF); Virus.
Figures
Similar articles
-
Associations between activation-induced cytidine deaminase/apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like cytidine deaminase expression, hepatitis B virus (HBV) replication and HBV-associated liver disease (Review).Mol Med Rep. 2015 Nov;12(5):6405-14. doi: 10.3892/mmr.2015.4312. Epub 2015 Sep 10. Mol Med Rep. 2015. PMID: 26398702 Free PMC article. Review.
-
Activation of TNF-α-AID axis and co-inhibitory signals in coordination with Th1-type immunity in a mouse model recapitulating hepatitis B.Antiviral Res. 2017 Mar;139:138-145. doi: 10.1016/j.antiviral.2017.01.004. Epub 2017 Jan 5. Antiviral Res. 2017. PMID: 28063995
-
Hepatitis B e Antigen Inhibits NF-κB Activity by Interrupting K63-Linked Ubiquitination of NEMO.J Virol. 2019 Jan 4;93(2):e00667-18. doi: 10.1128/JVI.00667-18. Print 2019 Jan 15. J Virol. 2019. PMID: 30404796 Free PMC article.
-
MafF Is an Antiviral Host Factor That Suppresses Transcription from Hepatitis B Virus Core Promoter.J Virol. 2021 Jul 12;95(15):e0076721. doi: 10.1128/JVI.00767-21. Epub 2021 Jul 12. J Virol. 2021. PMID: 33980595 Free PMC article.
-
Hepatitis B: modern concepts in pathogenesis--APOBEC3 cytidine deaminases as effectors in innate immunity against the hepatitis B virus.Curr Opin Infect Dis. 2008 Jun;21(3):298-303. doi: 10.1097/QCO.0b013e3282fe1bb2. Curr Opin Infect Dis. 2008. PMID: 18448976 Review.
Cited by
-
The off-target effects of AID in carcinogenesis.Front Immunol. 2023 Aug 4;14:1221528. doi: 10.3389/fimmu.2023.1221528. eCollection 2023. Front Immunol. 2023. PMID: 37600817 Free PMC article. Review.
-
The HBV Core Protein and Core Particle Both Bind to the PPiase Par14 and Par17 to Enhance Their Stabilities and HBV Replication.Front Microbiol. 2021 Dec 14;12:795047. doi: 10.3389/fmicb.2021.795047. eCollection 2021. Front Microbiol. 2021. PMID: 34970249 Free PMC article.
-
Expression and subcellular localisation of AID and APOBEC3 in adenoid and palatine tonsils.Sci Rep. 2018 Jan 17;8(1):918. doi: 10.1038/s41598-017-18732-w. Sci Rep. 2018. PMID: 29343743 Free PMC article.
-
Vitamin D signaling inhibits HBV activity by directly targeting the HBV core promoter.J Biol Chem. 2021 Oct;297(4):101233. doi: 10.1016/j.jbc.2021.101233. Epub 2021 Sep 23. J Biol Chem. 2021. PMID: 34562448 Free PMC article.
-
Serum IL-1β predicts de novo hepatitis B virus reactivation during direct-acting antiviral therapy for hepatitis C, not during anti-cancer/immunosuppressive therapy.Sci Rep. 2022 Oct 7;12(1):16800. doi: 10.1038/s41598-022-21315-z. Sci Rep. 2022. PMID: 36207368 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
