

Reactive oxygen species in inflammation and tissue injury
- PMID: 23991888
- PMCID: PMC3929010
- DOI: 10.1089/ars.2012.5149
Reactive oxygen species in inflammation and tissue injury
Abstract
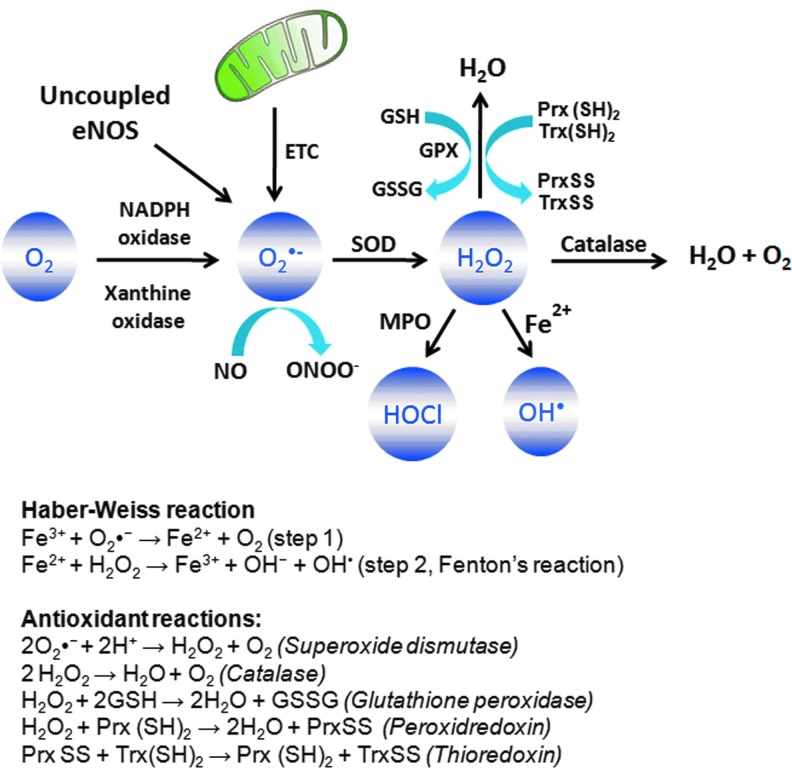
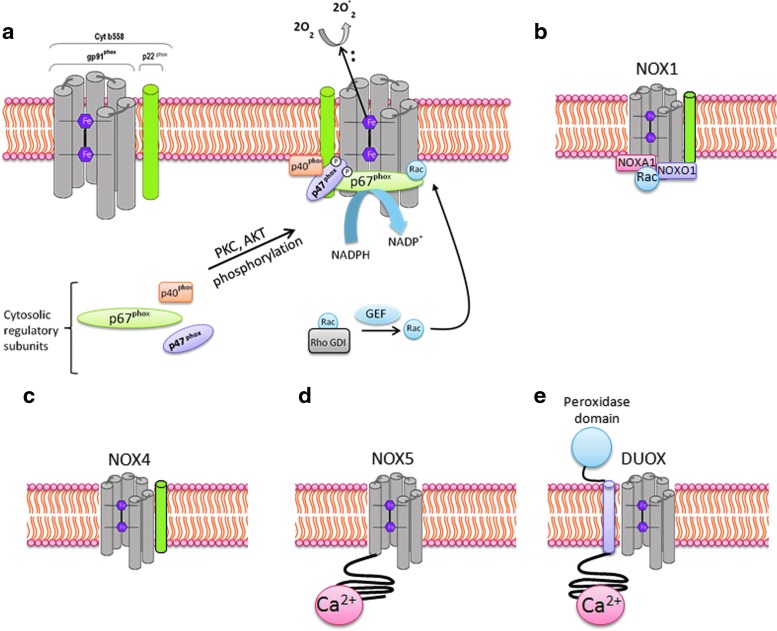
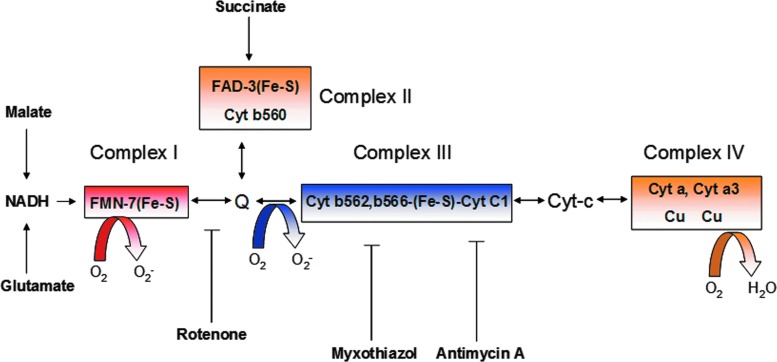
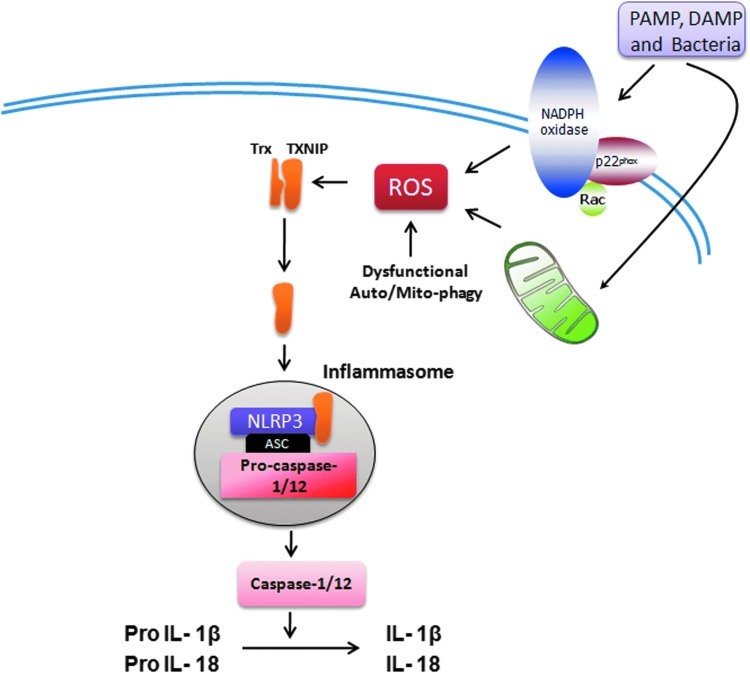
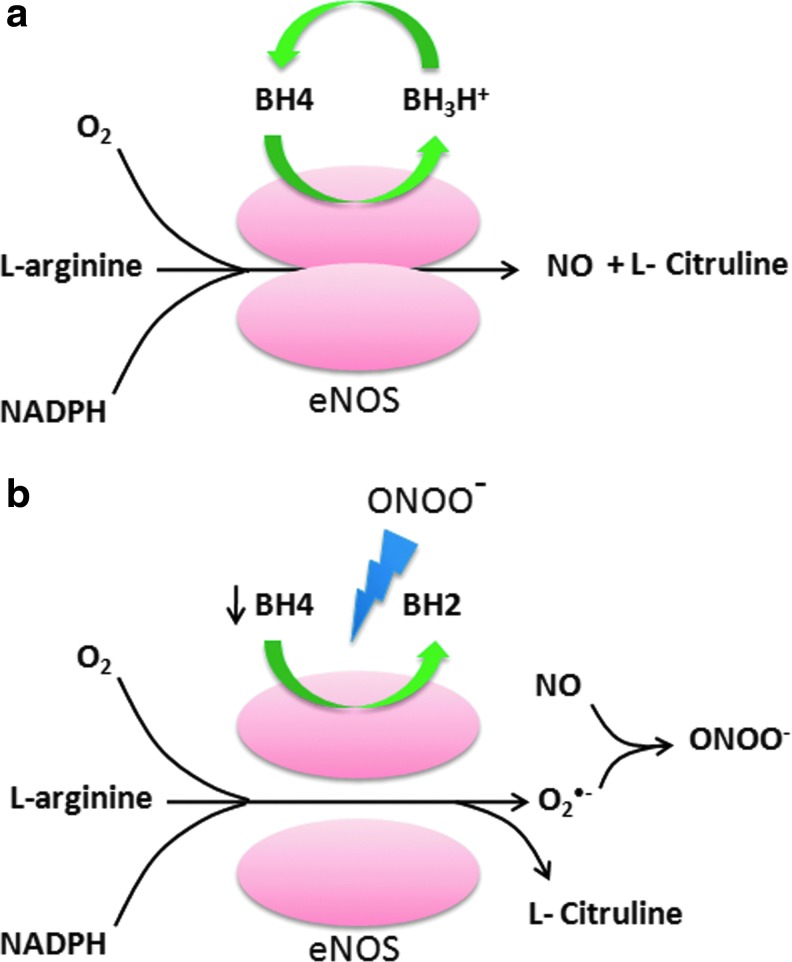
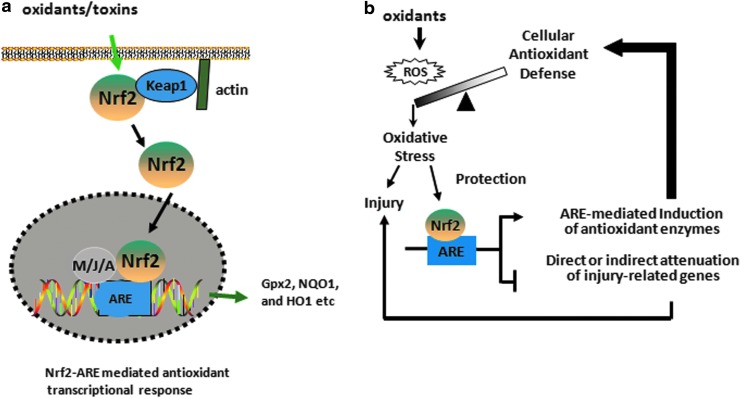
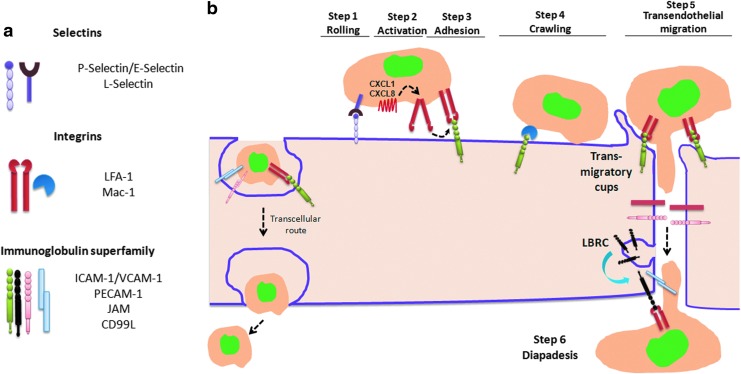
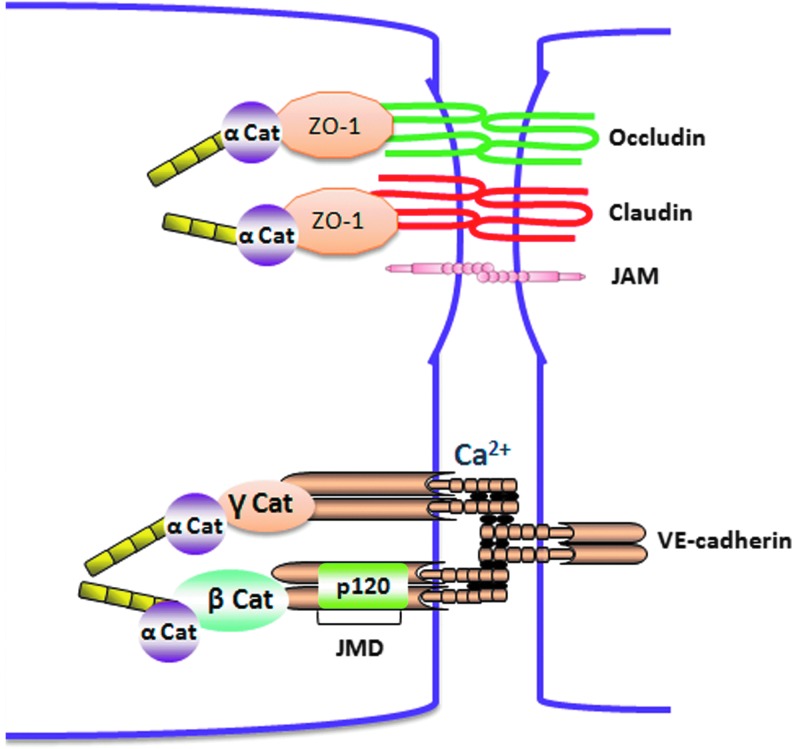
Abstract Reactive oxygen species (ROS) are key signaling molecules that play an important role in the progression of inflammatory disorders. An enhanced ROS generation by polymorphonuclear neutrophils (PMNs) at the site of inflammation causes endothelial dysfunction and tissue injury. The vascular endothelium plays an important role in passage of macromolecules and inflammatory cells from the blood to tissue. Under the inflammatory conditions, oxidative stress produced by PMNs leads to the opening of inter-endothelial junctions and promotes the migration of inflammatory cells across the endothelial barrier. The migrated inflammatory cells not only help in the clearance of pathogens and foreign particles but also lead to tissue injury. The current review compiles the past and current research in the area of inflammation with particular emphasis on oxidative stress-mediated signaling mechanisms that are involved in inflammation and tissue injury.
Figures

Similar articles
-
Causation by Diesel Exhaust Particles of Endothelial Dysfunctions in Cytotoxicity, Pro-inflammation, Permeability, and Apoptosis Induced by ROS Generation.Cardiovasc Toxicol. 2017 Oct;17(4):384-392. doi: 10.1007/s12012-016-9364-0. Cardiovasc Toxicol. 2017. PMID: 26965709 Review.
-
Endothelial cell dysfunction, injury and death.Handb Exp Pharmacol. 2006;(176 Pt 2):135-56. doi: 10.1007/3-540-36028-x_5. Handb Exp Pharmacol. 2006. PMID: 16999227 Review.
-
Reactive oxygen species formation by polymorphonuclear cells and mononuclear cells as a risk factor of cardiovascular diseases.Curr Pharm Biotechnol. 2006 Apr;7(2):73-80. doi: 10.2174/138920106776597612. Curr Pharm Biotechnol. 2006. PMID: 16724940 Review.
-
Interactions of oxidative stress and neurovascular inflammation in the pathogenesis of traumatic brain injury.Mol Neurobiol. 2015;51(3):966-79. doi: 10.1007/s12035-014-8752-3. Epub 2014 May 28. Mol Neurobiol. 2015. PMID: 24865512 Free PMC article. Review.
-
The interactions of oxidative stress and inflammation with vascular dysfunction in ageing: the vascular health triad.Age (Dordr). 2013 Jun;35(3):705-18. doi: 10.1007/s11357-012-9402-1. Epub 2012 Mar 28. Age (Dordr). 2013. PMID: 22453933 Free PMC article. Review.
Cited by
-
Effect of (R)-(-)-Linalool on endothelial damage: Sex differences.Biochem Biophys Rep. 2024 Oct 15;40:101846. doi: 10.1016/j.bbrep.2024.101846. eCollection 2024 Dec. Biochem Biophys Rep. 2024. PMID: 39483177 Free PMC article.
-
Overview of oxidative stress and inflammation in diabetes.J Diabetes. 2024 Oct;16(10):e70014. doi: 10.1111/1753-0407.70014. J Diabetes. 2024. PMID: 39435991 Free PMC article. Review.
-
KEAP1-NRF2 protein-protein interaction inhibitors: Design, pharmacological properties and therapeutic potential.Med Res Rev. 2023 Jan;43(1):237-287. doi: 10.1002/med.21925. Epub 2022 Sep 10. Med Res Rev. 2023. PMID: 36086898 Free PMC article. Review.
-
Development and validation of chest CT-based imaging biomarkers for early stage COVID-19 screening.Front Public Health. 2022 Sep 21;10:1004117. doi: 10.3389/fpubh.2022.1004117. eCollection 2022. Front Public Health. 2022. PMID: 36211676 Free PMC article.
-
Comparison of Anti-Oxidant and Anti-Inflammatory Effects between Fresh and Aged Black Garlic Extracts.Molecules. 2016 Mar 30;21(4):430. doi: 10.3390/molecules21040430. Molecules. 2016. PMID: 27043510 Free PMC article.
References
-
- Abid MR, Schoots IG, Spokes KC, Wu SQ, Mawhinney C, and Aird WC. Vascular endothelial growth factor-mediated induction of manganese superoxide dismutase occurs through redox-dependent regulation of forkhead and IkappaB/NF-kappaB. J Biol Chem 279: 44030–44038, 2004 - PubMed
-
- Aiello LP, Bursell SE, Clermont A, Duh E, Ishii H, Takagi C, Mori F, Ciulla TA, Ways K, Jirousek M, Smith LE, and King GL. Vascular endothelial growth factor-induced retinal permeability is mediated by protein kinase C in vivo and suppressed by an orally effective beta-isoform-selective inhibitor. Diabetes 46: 1473–1480, 1997 - PubMed
-
- Aikawa M, Sugiyama S, Hill CC, Voglic SJ, Rabkin E, Fukumoto Y, Schoen FJ, Witztum JL, and Libby P. Lipid lowering reduces oxidative stress and endothelial cell activation in rabbit atheroma. Circulation 106: 1390–1396, 2002 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
