

A creature with a hundred waggly tails: intrinsically disordered proteins in the ribosome
- PMID: 23942625
- PMCID: PMC7079807
- DOI: 10.1007/s00018-013-1446-6
A creature with a hundred waggly tails: intrinsically disordered proteins in the ribosome
Abstract
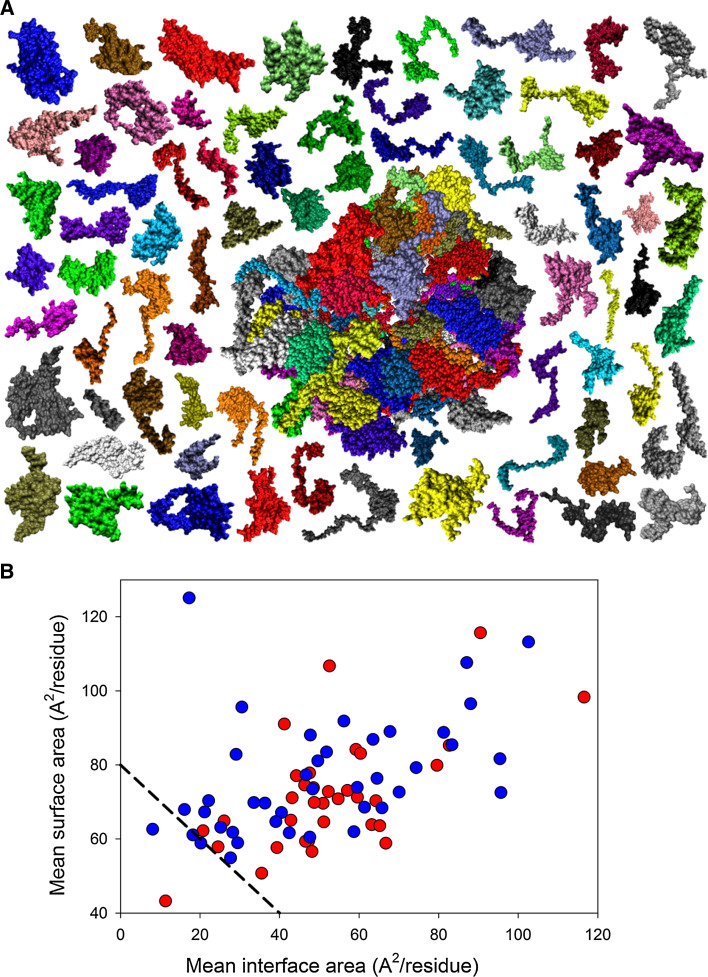
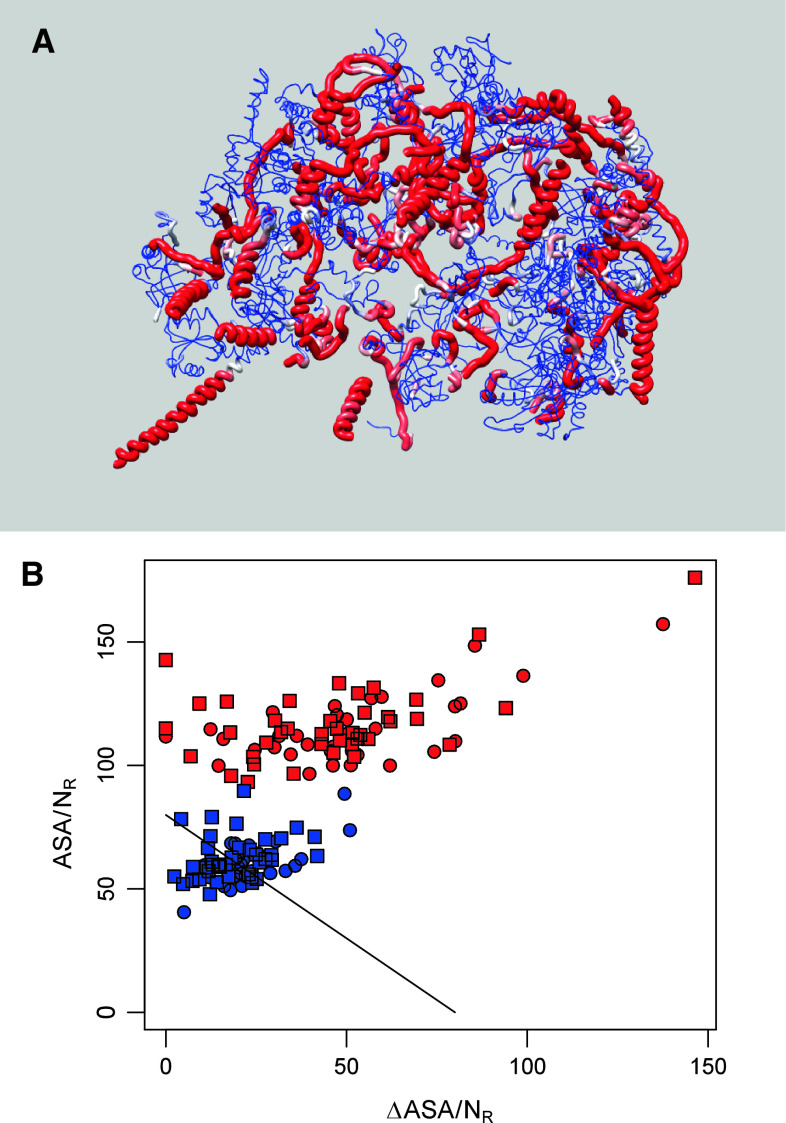
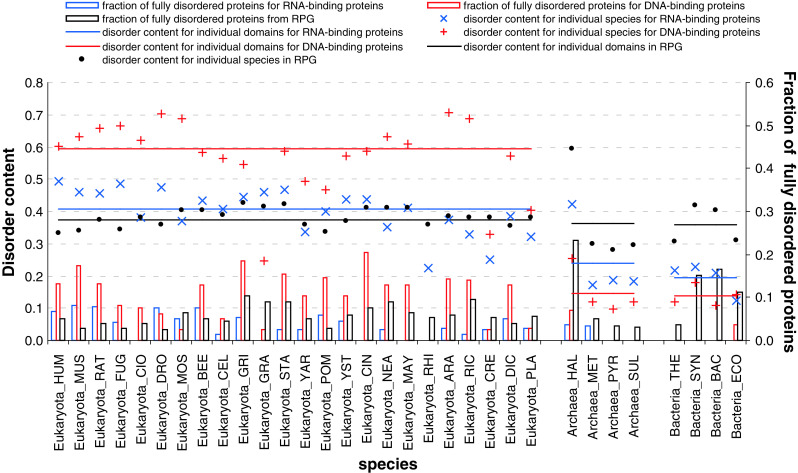
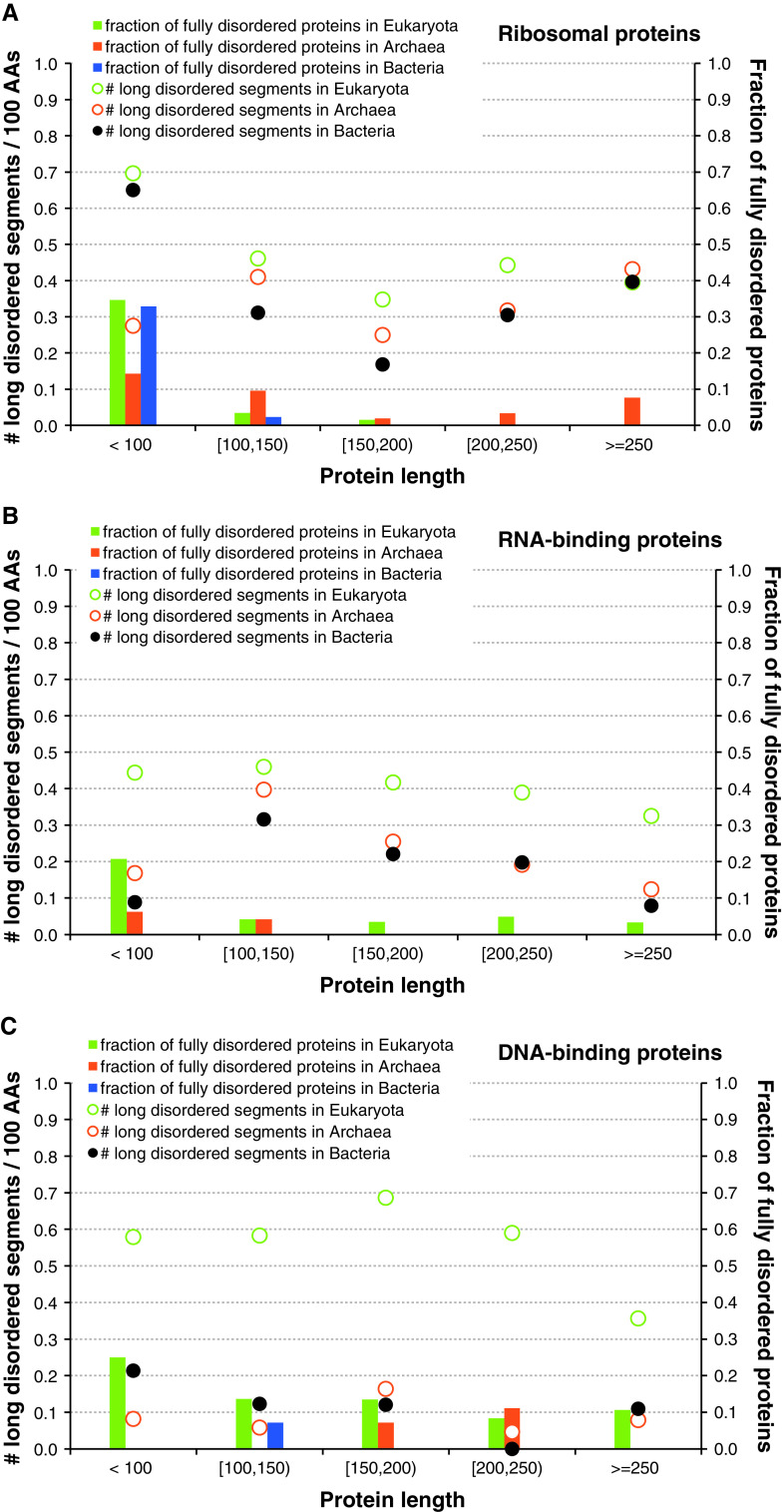
Intrinsic disorder (i.e., lack of a unique 3-D structure) is a common phenomenon, and many biologically active proteins are disordered as a whole, or contain long disordered regions. These intrinsically disordered proteins/regions constitute a significant part of all proteomes, and their functional repertoire is complementary to functions of ordered proteins. In fact, intrinsic disorder represents an important driving force for many specific functions. An illustrative example of such disorder-centric functional class is RNA-binding proteins. In this study, we present the results of comprehensive bioinformatics analyses of the abundance and roles of intrinsic disorder in 3,411 ribosomal proteins from 32 species. We show that many ribosomal proteins are intrinsically disordered or hybrid proteins that contain ordered and disordered domains. Predicted globular domains of many ribosomal proteins contain noticeable regions of intrinsic disorder. We also show that disorder in ribosomal proteins has different characteristics compared to other proteins that interact with RNA and DNA including overall abundance, evolutionary conservation, and involvement in protein-protein interactions. Furthermore, intrinsic disorder is not only abundant in the ribosomal proteins, but we demonstrate that it is absolutely necessary for their various functions.
Figures



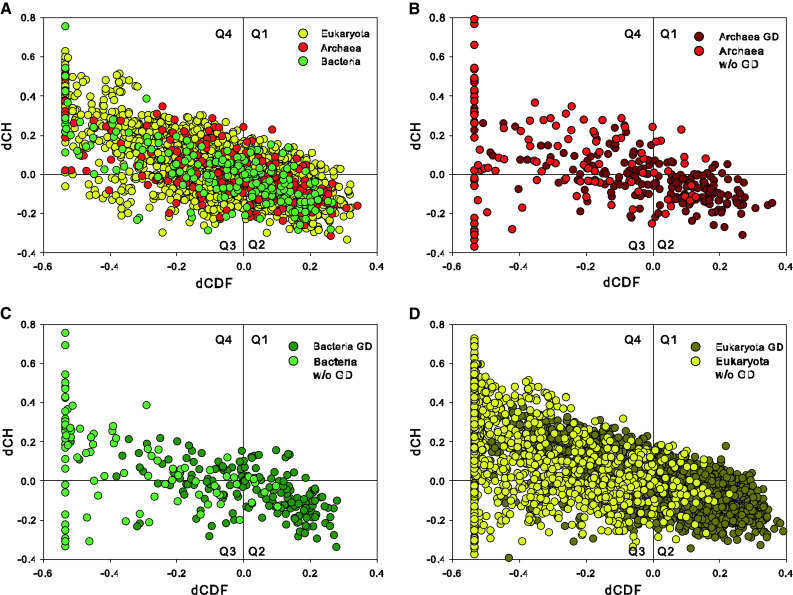
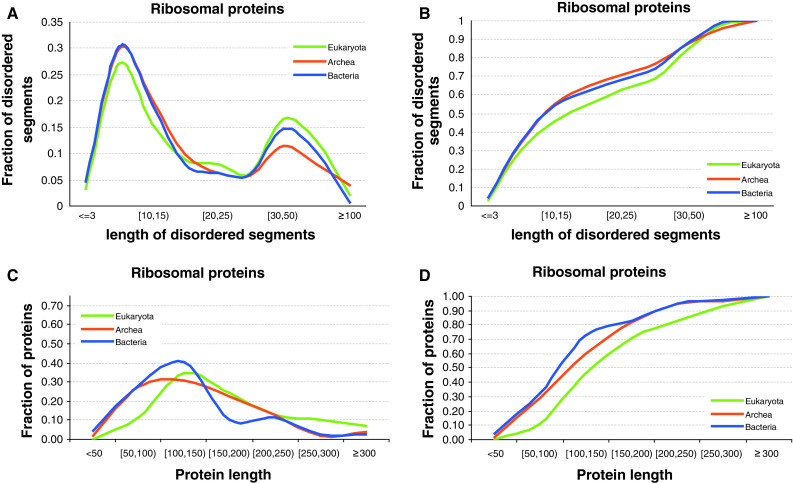



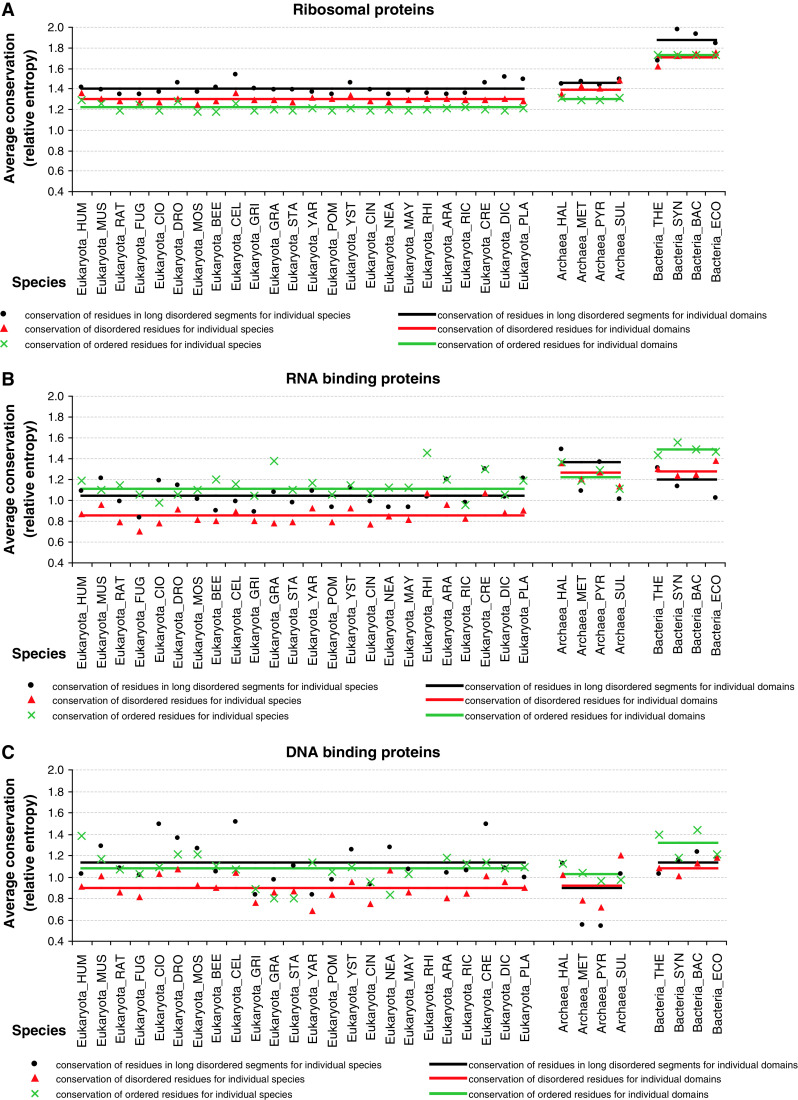
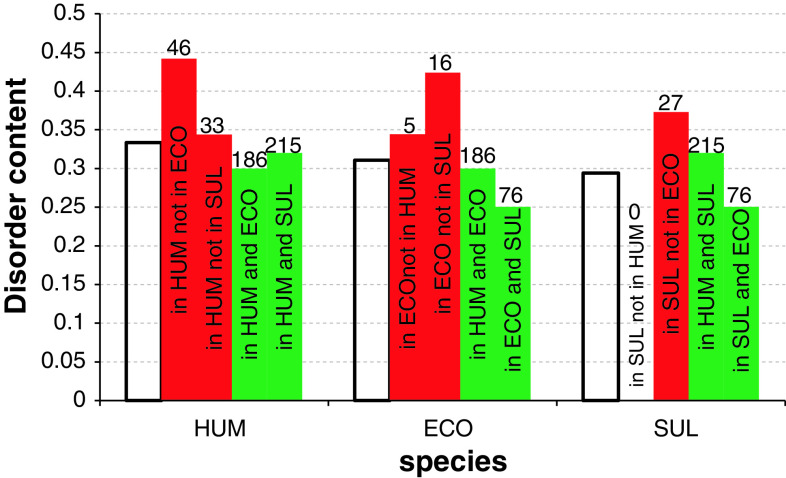
Similar articles
-
Conservation of intrinsic disorder in protein domains and families: II. functions of conserved disorder.J Proteome Res. 2006 Apr;5(4):888-98. doi: 10.1021/pr060049p. J Proteome Res. 2006. PMID: 16602696 Free PMC article.
-
Exceptionally abundant exceptions: comprehensive characterization of intrinsic disorder in all domains of life.Cell Mol Life Sci. 2015 Jan;72(1):137-51. doi: 10.1007/s00018-014-1661-9. Epub 2014 Jun 18. Cell Mol Life Sci. 2015. PMID: 24939692 Free PMC article.
-
Disordered nucleiome: Abundance of intrinsic disorder in the DNA- and RNA-binding proteins in 1121 species from Eukaryota, Bacteria and Archaea.Proteomics. 2016 May;16(10):1486-98. doi: 10.1002/pmic.201500177. Epub 2016 Apr 27. Proteomics. 2016. PMID: 27037624
-
Ribosomal proteins: structure, function, and evolution.Biochemistry (Mosc). 2012 Jun;77(6):562-74. doi: 10.1134/S0006297912060028. Biochemistry (Mosc). 2012. PMID: 22817455 Review.
-
Functional unfoldomics: Roles of intrinsic disorder in protein (multi)functionality.Adv Protein Chem Struct Biol. 2024;138:179-210. doi: 10.1016/bs.apcsb.2023.11.001. Epub 2023 Nov 22. Adv Protein Chem Struct Biol. 2024. PMID: 38220424 Review.
Cited by
-
Mechanistic insights into mammalian stress granule dynamics.J Cell Biol. 2016 Nov 7;215(3):313-323. doi: 10.1083/jcb.201609081. J Cell Biol. 2016. PMID: 27821493 Free PMC article. Review.
-
Structural Basis of the Subcellular Topology Landscape of Escherichia coli.Front Microbiol. 2019 Jul 24;10:1670. doi: 10.3389/fmicb.2019.01670. eCollection 2019. Front Microbiol. 2019. PMID: 31404336 Free PMC article.
-
Sequence-Derived Markers of Drug Targets and Potentially Druggable Human Proteins.Front Genet. 2019 Nov 15;10:1075. doi: 10.3389/fgene.2019.01075. eCollection 2019. Front Genet. 2019. PMID: 31803227 Free PMC article.
-
Lighting up Nobel Prize-winning studies with protein intrinsic disorder.Cell Mol Life Sci. 2022 Jul 26;79(8):449. doi: 10.1007/s00018-022-04468-y. Cell Mol Life Sci. 2022. PMID: 35882686 Free PMC article. Review.
-
The RNA-Binding Function of Ribosomal Proteins and Ribosome Biogenesis Factors in Human Health and Disease.Biomedicines. 2023 Nov 4;11(11):2969. doi: 10.3390/biomedicines11112969. Biomedicines. 2023. PMID: 38001969 Free PMC article. Review.
References
-
- Dunker AK, Obradovic Z, Romero P, Garner EC, Brown CJ. Intrinsic protein disorder in complete genomes. Genome Inf Ser Workshop Genome Inf. 2000;11:161–171. - PubMed
-
- Ward JJ, Sodhi JS, McGuffin LJ, Buxton BF, Jones DT. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J Mol Biol. 2004;337(3):635–645. - PubMed
-
- Uversky VN, Gillespie JR, Fink AL. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins. 2000;41(3):415–427. - PubMed
-
- Xue B, Dunker AK, Uversky VN. Orderly order in protein intrinsic disorder distribution: disorder in 3500 proteomes from viruses and the three domains of life. J Biomol Struct Dyn. 2012;30(2):137–149. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
