

The genome sequence of the colonial chordate, Botryllus schlosseri
- PMID: 23840927
- PMCID: PMC3699833
- DOI: 10.7554/eLife.00569
The genome sequence of the colonial chordate, Botryllus schlosseri
Abstract
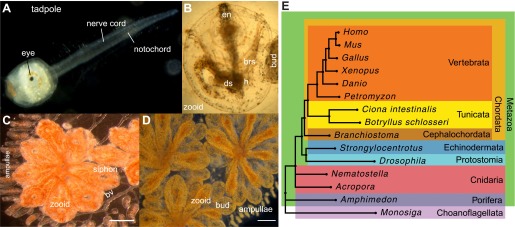
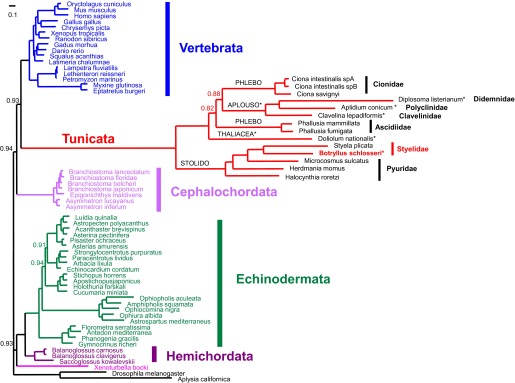
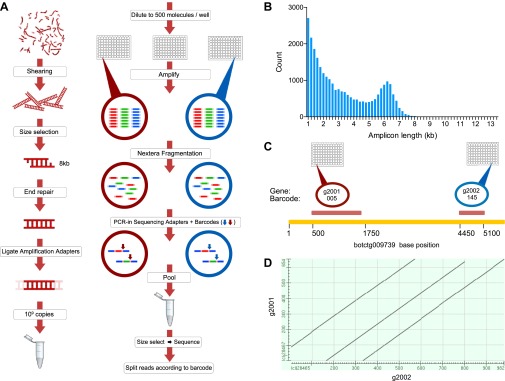
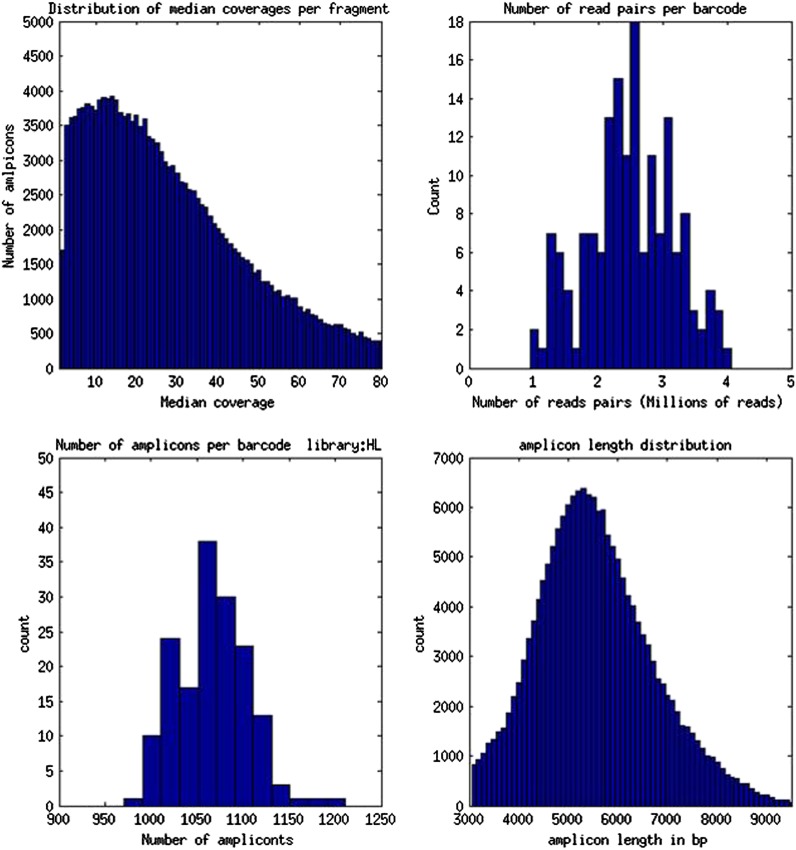
Botryllus schlosseri is a colonial urochordate that follows the chordate plan of development following sexual reproduction, but invokes a stem cell-mediated budding program during subsequent rounds of asexual reproduction. As urochordates are considered to be the closest living invertebrate relatives of vertebrates, they are ideal subjects for whole genome sequence analyses. Using a novel method for high-throughput sequencing of eukaryotic genomes, we sequenced and assembled 580 Mbp of the B. schlosseri genome. The genome assembly is comprised of nearly 14,000 intron-containing predicted genes, and 13,500 intron-less predicted genes, 40% of which could be confidently parceled into 13 (of 16 haploid) chromosomes. A comparison of homologous genes between B. schlosseri and other diverse taxonomic groups revealed genomic events underlying the evolution of vertebrates and lymphoid-mediated immunity. The B. schlosseri genome is a community resource for studying alternative modes of reproduction, natural transplantation reactions, and stem cell-mediated regeneration. DOI:http://dx.doi.org/10.7554/eLife.00569.001.
Keywords: Botryllus schlosseri; Other; genome; hematopoiesis; stem cell; tunicates; vertebrate evolution.
Conflict of interest statement
Stanford has filed US and International patent application numbers 61/532,882 and 13/608,778 entitled “Methods for obtaining a sequence” with inventors AV, DP, and SRQ. DP and SRQ are co-founders of Moleculo Inc.
The other authors declare that no competing interests exist.
Figures

Similar articles
-
Botryllus schlosseri as a Unique Colonial Chordate Model for the Study and Modulation of Innate Immune Activity.Mar Drugs. 2021 Aug 9;19(8):454. doi: 10.3390/md19080454. Mar Drugs. 2021. PMID: 34436293 Free PMC article. Review.
-
Stem cell-mediated development, regeneration, chimerism, and aging in the colonial chordate Botryllus schlosseri.Genesis. 2023 Nov;61(6):e23542. doi: 10.1002/dvg.23542. Epub 2023 Oct 27. Genesis. 2023. PMID: 37888861 Free PMC article.
-
Characterization of Ambra1 in asexual cycle of a non-vertebrate chordate, the colonial tunicate Botryllus schlosseri, and phylogenetic analysis of the protein group in Bilateria.Mol Phylogenet Evol. 2016 Feb;95:46-57. doi: 10.1016/j.ympev.2015.11.001. Epub 2015 Nov 23. Mol Phylogenet Evol. 2016. PMID: 26611831
-
Botryllus schlosseri, an emerging model for the study of aging, stem cells, and mechanisms of regeneration.Invertebr Reprod Dev. 2015 Jan 30;59(sup1):33-38. doi: 10.1080/07924259.2014.944673. Epub 2014 Dec 9. Invertebr Reprod Dev. 2015. PMID: 26136618 Free PMC article.
-
Evolutionary perspective on the hematopoietic system through a colonial chordate: allogeneic immunity and hematopoiesis.Curr Opin Immunol. 2020 Feb;62:91-98. doi: 10.1016/j.coi.2019.12.006. Epub 2020 Jan 16. Curr Opin Immunol. 2020. PMID: 31954962 Free PMC article. Review.
Cited by
-
Contributions from both the brain and the vascular network guide behavior in the colonial tunicate Botryllus schlosseri.J Exp Biol. 2022 Nov 15;225(22):jeb244491. doi: 10.1242/jeb.244491. Epub 2022 Nov 25. J Exp Biol. 2022. PMID: 36314197 Free PMC article.
-
Rapid Evolutionary Rates and Unique Genomic Signatures Discovered in the First Reference Genome for the Southern Ocean Salp, Salpa thompsoni (Urochordata, Thaliacea).Genome Biol Evol. 2016 Oct 30;8(10):3171-3186. doi: 10.1093/gbe/evw215. Genome Biol Evol. 2016. PMID: 27624472 Free PMC article.
-
The genetic basis for ecological adaptation of the Atlantic herring revealed by genome sequencing.Elife. 2016 May 3;5:e12081. doi: 10.7554/eLife.12081. Elife. 2016. PMID: 27138043 Free PMC article.
-
Illumina Synthetic Long Read Sequencing Allows Recovery of Missing Sequences even in the "Finished" C. elegans Genome.Sci Rep. 2015 Jun 3;5:10814. doi: 10.1038/srep10814. Sci Rep. 2015. PMID: 26039588 Free PMC article.
-
Haplotype-resolved genome sequencing: experimental methods and applications.Nat Rev Genet. 2015 Jun;16(6):344-58. doi: 10.1038/nrg3903. Epub 2015 May 7. Nat Rev Genet. 2015. PMID: 25948246 Review.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
