

Sirt3 regulates metabolic flexibility of skeletal muscle through reversible enzymatic deacetylation
- PMID: 23835326
- PMCID: PMC3781465
- DOI: 10.2337/db12-1650
Sirt3 regulates metabolic flexibility of skeletal muscle through reversible enzymatic deacetylation
Abstract
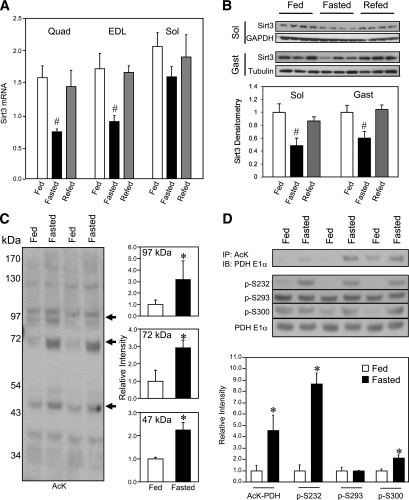
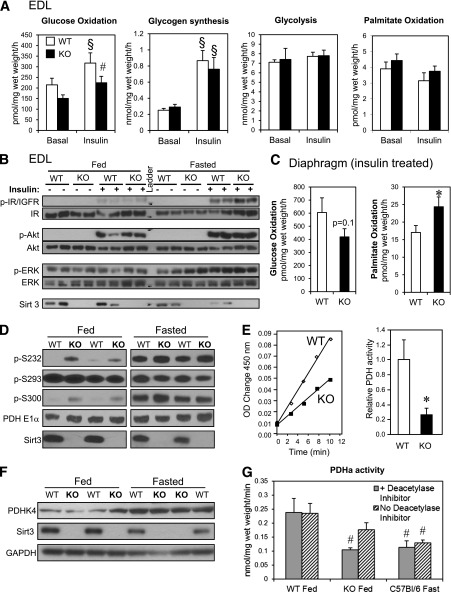
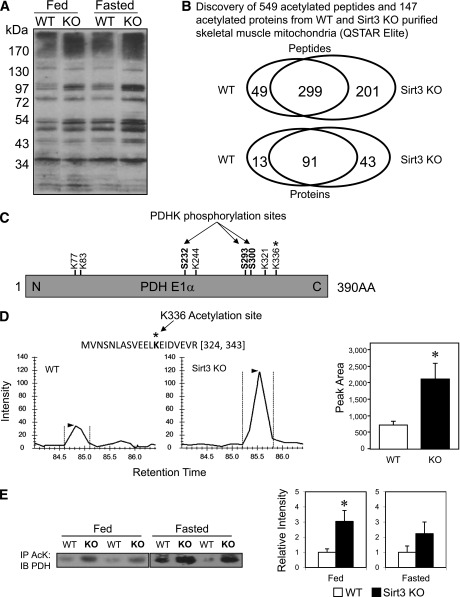
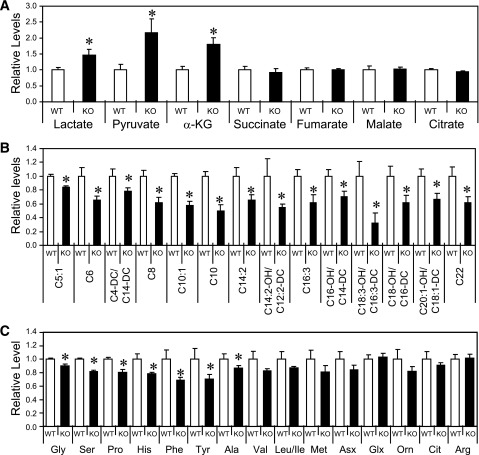
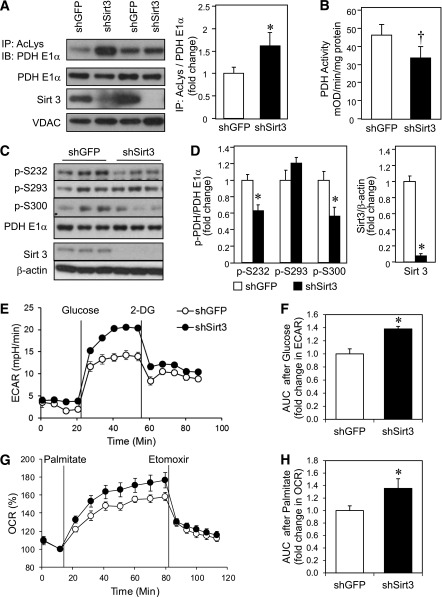
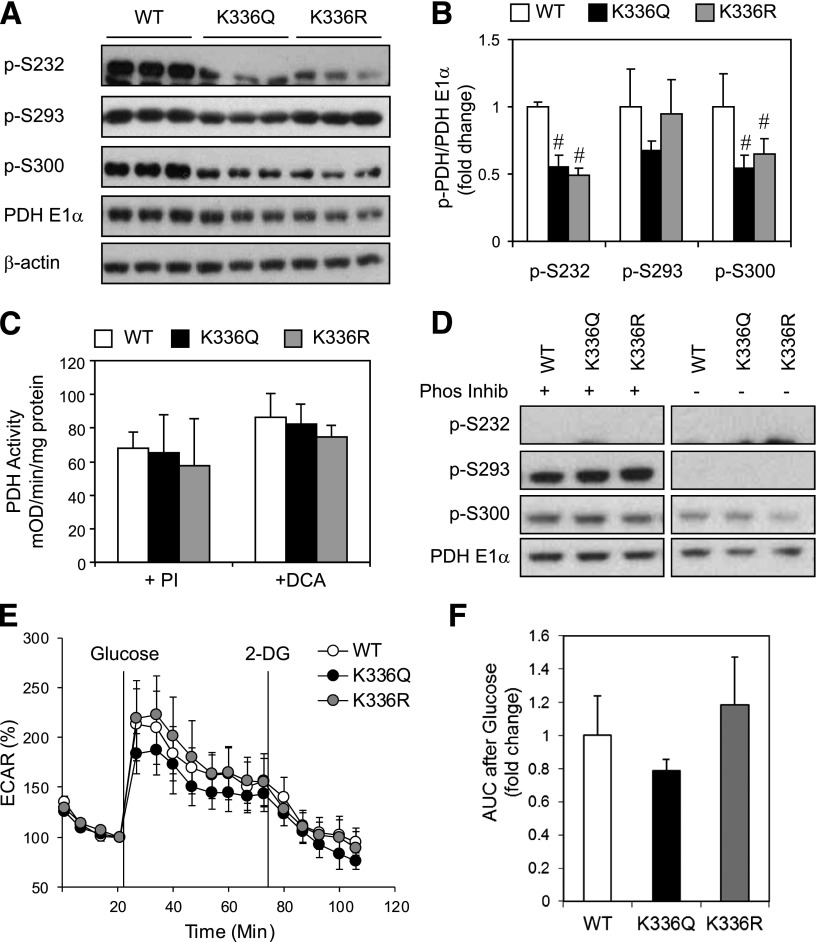
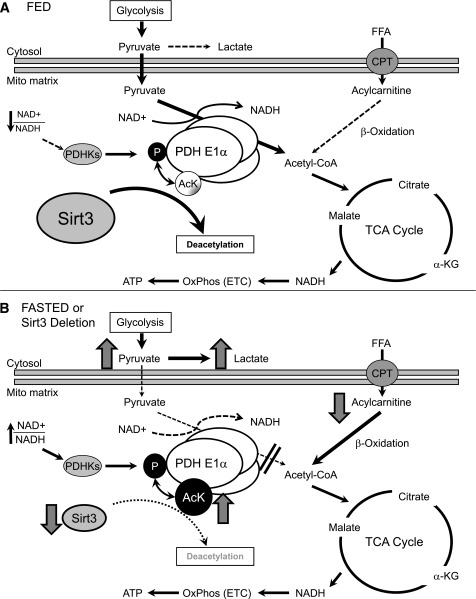
Sirt3 is an NAD(+)-dependent deacetylase that regulates mitochondrial function by targeting metabolic enzymes and proteins. In fasting mice, Sirt3 expression is decreased in skeletal muscle resulting in increased mitochondrial protein acetylation. Deletion of Sirt3 led to impaired glucose oxidation in muscle, which was associated with decreased pyruvate dehydrogenase (PDH) activity, accumulation of pyruvate and lactate metabolites, and an inability of insulin to suppress fatty acid oxidation. Antibody-based acetyl-peptide enrichment and mass spectrometry of mitochondrial lysates from WT and Sirt3 KO skeletal muscle revealed that a major target of Sirt3 deacetylation is the E1α subunit of PDH (PDH E1α). Sirt3 knockout in vivo and Sirt3 knockdown in myoblasts in vitro induced hyperacetylation of the PDH E1α subunit, altering its phosphorylation leading to suppressed PDH enzymatic activity. The inhibition of PDH activity resulting from reduced levels of Sirt3 induces a switch of skeletal muscle substrate utilization from carbohydrate oxidation toward lactate production and fatty acid utilization even in the fed state, contributing to a loss of metabolic flexibility. Thus, Sirt3 plays an important role in skeletal muscle mitochondrial substrate choice and metabolic flexibility in part by regulating PDH function through deacetylation.
Figures
Similar articles
-
PGC-1α and fasting-induced PDH regulation in mouse skeletal muscle.Physiol Rep. 2017 Apr;5(7):e13222. doi: 10.14814/phy2.13222. Physiol Rep. 2017. PMID: 28400503 Free PMC article.
-
MicroRNA-195 Regulates Metabolism in Failing Myocardium Via Alterations in Sirtuin 3 Expression and Mitochondrial Protein Acetylation.Circulation. 2018 May 8;137(19):2052-2067. doi: 10.1161/CIRCULATIONAHA.117.030486. Epub 2018 Jan 12. Circulation. 2018. PMID: 29330215 Free PMC article.
-
SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation.Nature. 2010 Mar 4;464(7285):121-5. doi: 10.1038/nature08778. Nature. 2010. PMID: 20203611 Free PMC article.
-
SIRT3 regulates mitochondrial protein acetylation and intermediary metabolism.Cold Spring Harb Symp Quant Biol. 2011;76:267-77. doi: 10.1101/sqb.2011.76.010850. Epub 2011 Nov 23. Cold Spring Harb Symp Quant Biol. 2011. PMID: 22114326 Review.
-
Acetylation of Mitochondrial Proteins in the Heart: The Role of SIRT3.Front Physiol. 2018 Aug 7;9:1094. doi: 10.3389/fphys.2018.01094. eCollection 2018. Front Physiol. 2018. PMID: 30131726 Free PMC article. Review.
Cited by
-
Nitrate-induced improvements in exercise performance are coincident with exuberant changes in metabolic genes and the metabolome in zebrafish (Danio rerio) skeletal muscle.J Appl Physiol (1985). 2021 Jul 1;131(1):142-157. doi: 10.1152/japplphysiol.00185.2021. Epub 2021 May 27. J Appl Physiol (1985). 2021. PMID: 34043471 Free PMC article.
-
Mitophagy plays a central role in mitochondrial ageing.Mamm Genome. 2016 Aug;27(7-8):381-95. doi: 10.1007/s00335-016-9651-x. Epub 2016 Jun 28. Mamm Genome. 2016. PMID: 27352213 Free PMC article. Review.
-
Mitochondrial sirtuins: Energy dynamics and cancer metabolism.Mol Cells. 2024 Feb;47(2):100029. doi: 10.1016/j.mocell.2024.100029. Epub 2024 Feb 6. Mol Cells. 2024. PMID: 38331199 Free PMC article. Review.
-
Lack of Skeletal Muscle IL-6 Affects Pyruvate Dehydrogenase Activity at Rest and during Prolonged Exercise.PLoS One. 2016 Jun 21;11(6):e0156460. doi: 10.1371/journal.pone.0156460. eCollection 2016. PLoS One. 2016. PMID: 27327080 Free PMC article.
-
The Role of Palmitoleic Acid in Regulating Hepatic Gluconeogenesis through SIRT3 in Obese Mice.Nutrients. 2022 Apr 1;14(7):1482. doi: 10.3390/nu14071482. Nutrients. 2022. PMID: 35406095 Free PMC article.
References
-
- Fueger PT, Hess HS, Bracy DP, et al. Regulation of insulin-stimulated muscle glucose uptake in the conscious mouse: role of glucose transport is dependent on glucose phosphorylation capacity. Endocrinology 2004;145:4912–4916 - PubMed
-
- Storlien L, Oakes ND, Kelley DE. Metabolic flexibility. Proc Nutr Soc 2004;63:363–368 - PubMed
-
- van Loon LJ. Use of intramuscular triacylglycerol as a substrate source during exercise in humans. J Appl Physiol 2004;97:1170–1187 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
