

SAD kinases sculpt axonal arbors of sensory neurons through long- and short-term responses to neurotrophin signals
- PMID: 23790753
- PMCID: PMC3725037
- DOI: 10.1016/j.neuron.2013.05.017
SAD kinases sculpt axonal arbors of sensory neurons through long- and short-term responses to neurotrophin signals
Abstract
Extrinsic cues activate intrinsic signaling mechanisms to pattern neuronal shape and connectivity. We showed previously that three cytoplasmic Ser/Thr kinases, LKB1, SAD-A, and SAD-B, control early axon-dendrite polarization in forebrain neurons. Here, we assess their role in other neuronal types. We found that all three kinases are dispensable for axon formation outside of the cortex but that SAD kinases are required for formation of central axonal arbors by subsets of sensory neurons. The requirement for SAD kinases is most prominent in NT-3 dependent neurons. SAD kinases transduce NT-3 signals in two ways through distinct pathways. First, sustained NT-3/TrkC signaling increases SAD protein levels. Second, short-duration NT-3/TrkC signals transiently activate SADs by inducing dephosphorylation of C-terminal domains, thereby allowing activating phosphorylation of the kinase domain. We propose that SAD kinases integrate long- and short-duration signals from extrinsic cues to sculpt axon arbors within the CNS.
Copyright © 2013 Elsevier Inc. All rights reserved.
Figures
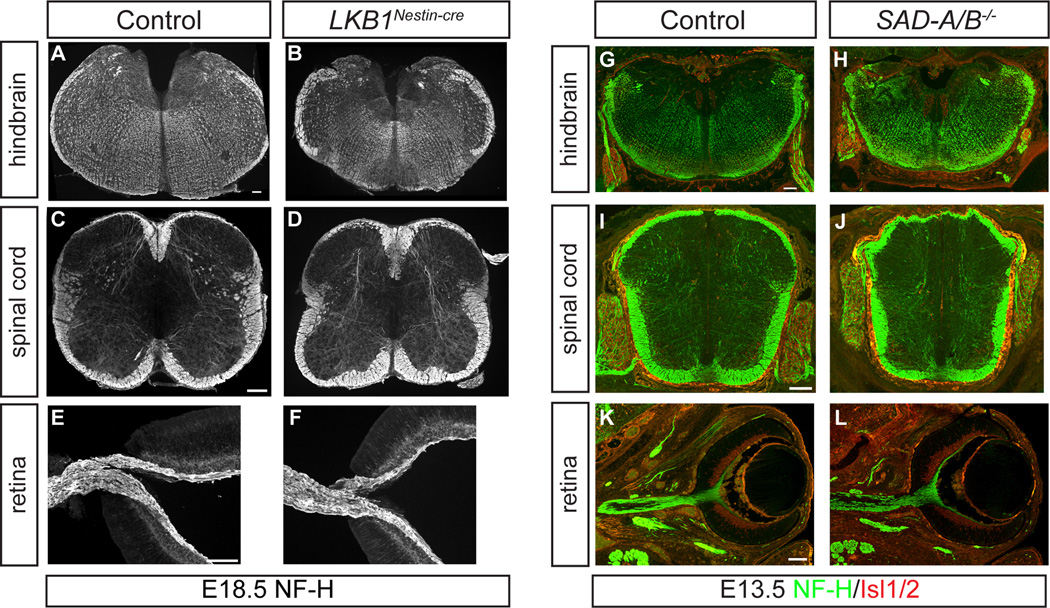
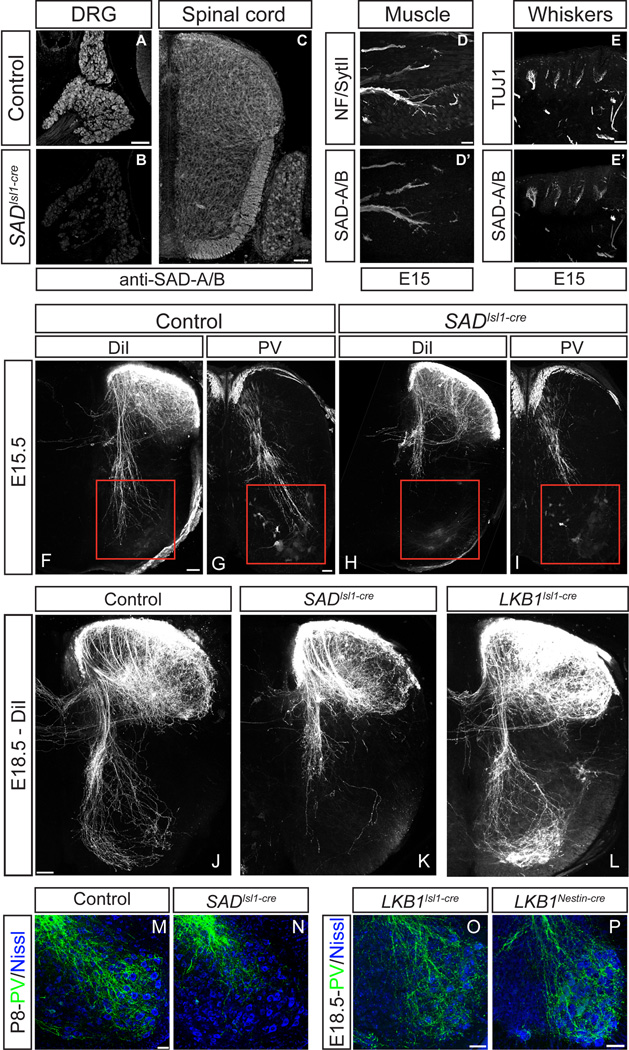
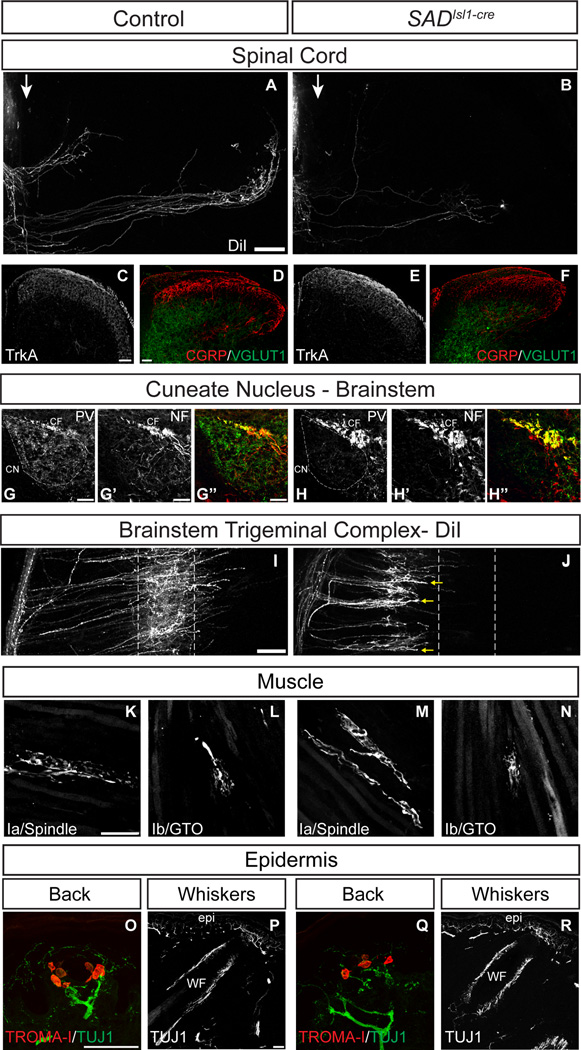
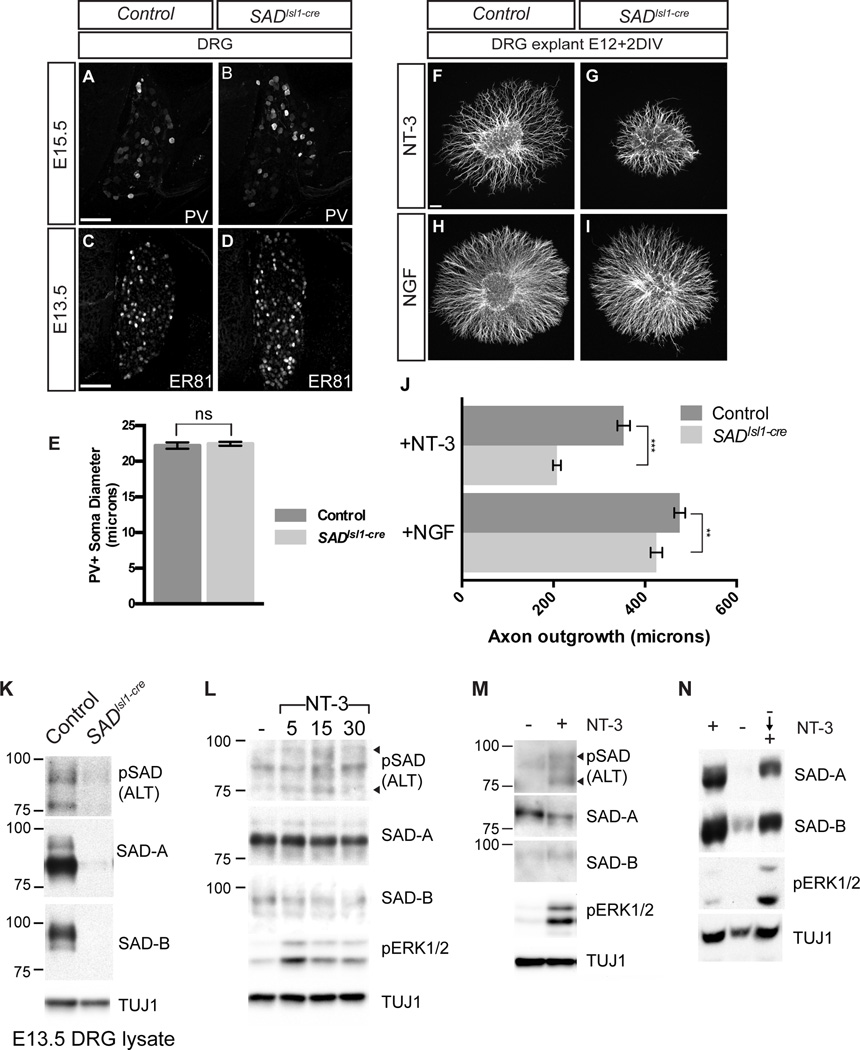

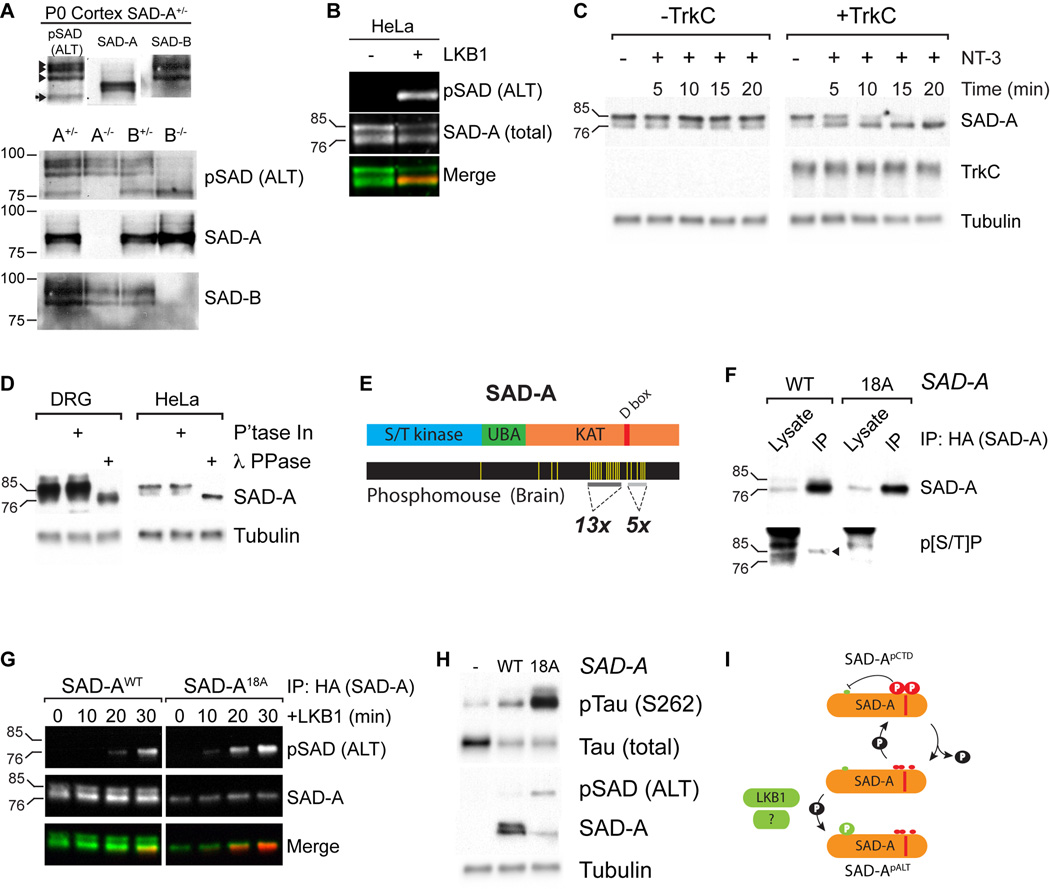

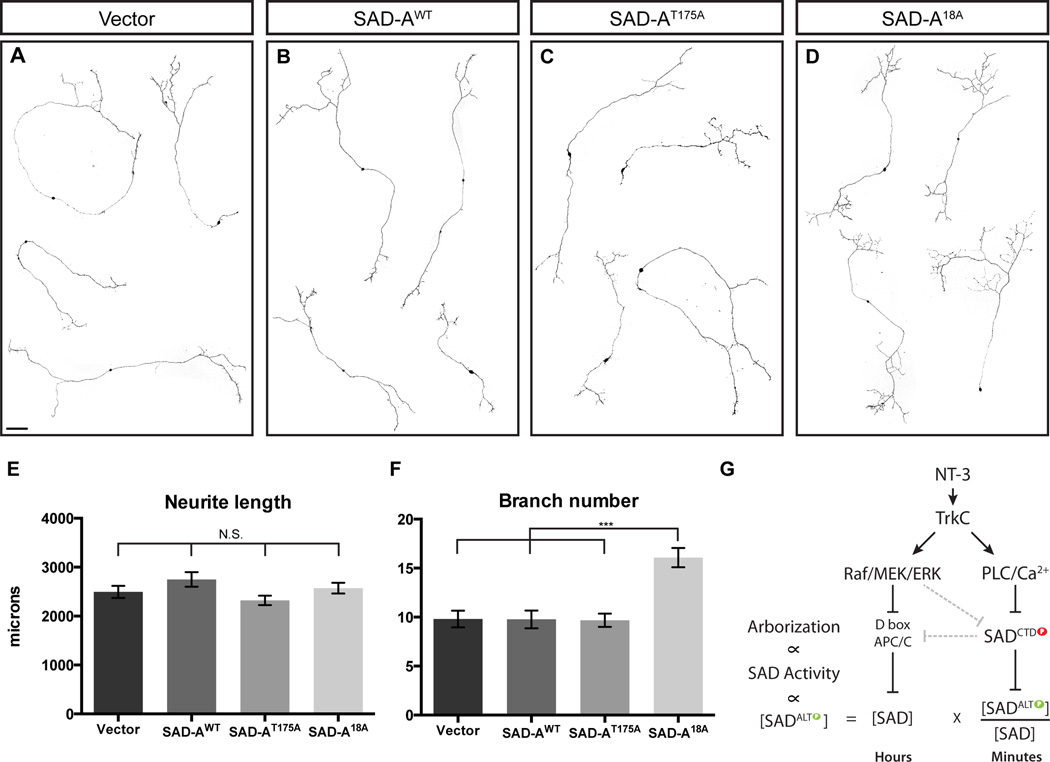
Comment in
-
Neuronal development: SAD kinases make happy axons.Curr Biol. 2013 Sep 9;23(17):R720-3. doi: 10.1016/j.cub.2013.07.073. Curr Biol. 2013. PMID: 24028951 Free PMC article.
Similar articles
-
Neuronal development: SAD kinases make happy axons.Curr Biol. 2013 Sep 9;23(17):R720-3. doi: 10.1016/j.cub.2013.07.073. Curr Biol. 2013. PMID: 24028951 Free PMC article.
-
LKB1 and SAD kinases define a pathway required for the polarization of cortical neurons.Cell. 2007 May 4;129(3):549-63. doi: 10.1016/j.cell.2007.03.025. Cell. 2007. PMID: 17482548
-
Role of LKB1-SAD/MARK pathway in neuronal polarization.Dev Neurobiol. 2011 Jun;71(6):508-27. doi: 10.1002/dneu.20884. Dev Neurobiol. 2011. PMID: 21416623 Review.
-
p75 neurotrophin receptor is implicated in the ability of neurotrophin-3 to negatively modulate activated ERK1/2 signaling in TrkA-expressing adult sensory neurons.J Comp Neurol. 2009 Sep 1;516(1):49-58. doi: 10.1002/cne.22098. J Comp Neurol. 2009. PMID: 19565663
-
[The role of TrkC receptor and neurotrophin 3 in the development and function of neural cells].Postepy Hig Med Dosw (Online). 2005;59:517-22. Postepy Hig Med Dosw (Online). 2005. PMID: 16258418 Review. Polish.
Cited by
-
The AMPK-related kinase NUAK1 controls cortical axons branching by locally modulating mitochondrial metabolic functions.Nat Commun. 2024 Mar 21;15(1):2487. doi: 10.1038/s41467-024-46146-6. Nat Commun. 2024. PMID: 38514619 Free PMC article.
-
Tail Nerve Electrical Stimulation and Electro-Acupuncture Can Protect Spinal Motor Neurons and Alleviate Muscle Atrophy after Spinal Cord Transection in Rats.Neural Plast. 2017;2017:7351238. doi: 10.1155/2017/7351238. Epub 2017 Jun 28. Neural Plast. 2017. PMID: 28744378 Free PMC article.
-
Liver Kinase B1 Functions as a Regulator for Neural Development and a Therapeutic Target for Neural Repair.Cells. 2022 Sep 14;11(18):2861. doi: 10.3390/cells11182861. Cells. 2022. PMID: 36139438 Free PMC article. Review.
-
SAD-A Promotes Glucose-Stimulated Insulin Secretion Through Phosphorylation and Inhibition of GDIα in Male Islet β Cells.Endocrinology. 2018 Aug 1;159(8):3036-3047. doi: 10.1210/en.2017-03243. Endocrinology. 2018. PMID: 29873699 Free PMC article.
-
An NT-3-releasing bioscaffold supports the formation of TrkC-modified neural stem cell-derived neural network tissue with efficacy in repairing spinal cord injury.Bioact Mater. 2021 Apr 7;6(11):3766-3781. doi: 10.1016/j.bioactmat.2021.03.036. eCollection 2021 Nov. Bioact Mater. 2021. PMID: 33898877 Free PMC article.
References
-
- Airaksinen MS, Koltzenburg M, Lewin GR, Masu Y, Helbig C, Wolf E, Brem G, Toyka KV, Thoenen H, Meyer M. Specific subtypes of cutaneous mechanoreceptors require neurotrophin-3 following peripheral target innervation. Neuron. 1996;16:287–295. - PubMed
-
- Alessi DR, Sakamoto K, Bayascas JR. LKB1-dependent signaling pathways. Annu Rev Biochem. 2006;75:137–163. - PubMed
-
- Alvarado-Kristensson M, Rodríguez MJ, Silió V, Valpuesta JM, Carrera AC. SADB phosphorylation of gamma-tubulin regulates centrosome duplication. Nat Cell Biol. 2009;11:1081–1092. - PubMed
-
- Arber S, Ladle DR, Lin JH, Frank E, Jessell TM. ETS gene Er81 controls the formation of functional connections between group Ia sensory afferents and motor neurons. Cell. 2000;101:485–498. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
