

Whole-genome sequencing identifies recurrent mutations in hepatocellular carcinoma
- PMID: 23788652
- PMCID: PMC3759719
- DOI: 10.1101/gr.154492.113
Whole-genome sequencing identifies recurrent mutations in hepatocellular carcinoma
Abstract
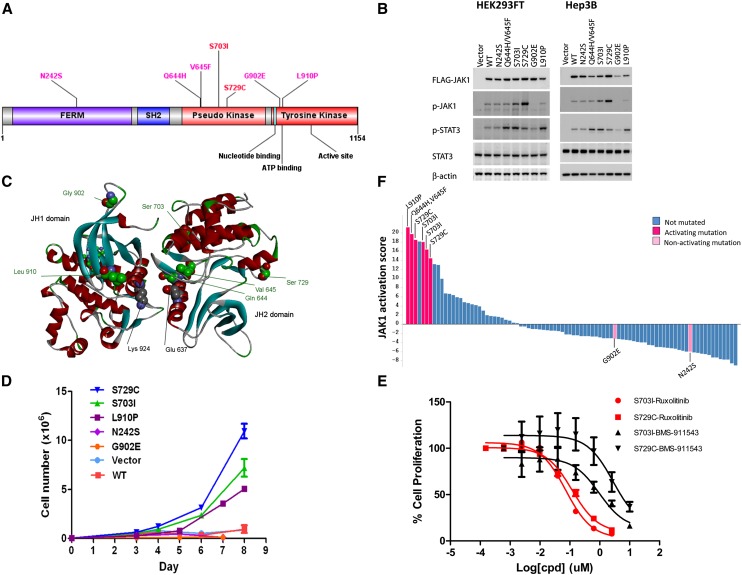
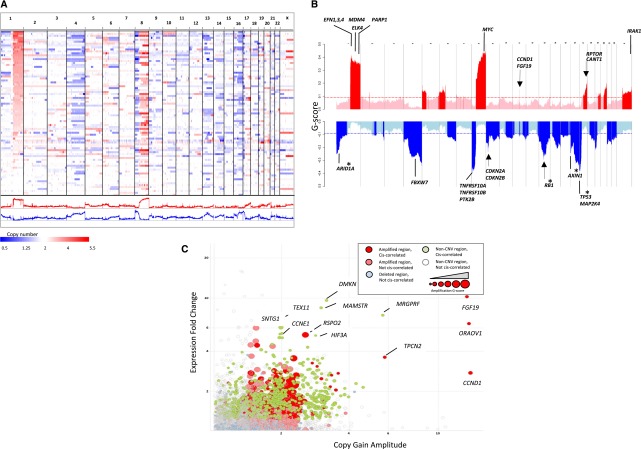
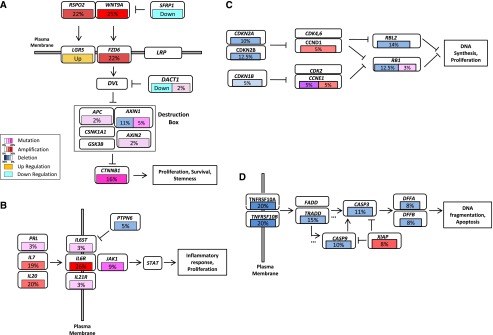
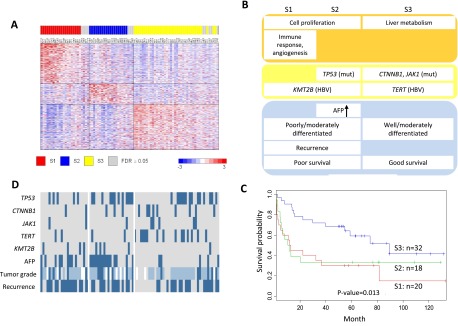
Hepatocellular carcinoma (HCC) is one of the most deadly cancers worldwide and has no effective treatment, yet the molecular basis of hepatocarcinogenesis remains largely unknown. Here we report findings from a whole-genome sequencing (WGS) study of 88 matched HCC tumor/normal pairs, 81 of which are Hepatitis B virus (HBV) positive, seeking to identify genetically altered genes and pathways implicated in HBV-associated HCC. We find beta-catenin to be the most frequently mutated oncogene (15.9%) and TP53 the most frequently mutated tumor suppressor (35.2%). The Wnt/beta-catenin and JAK/STAT pathways, altered in 62.5% and 45.5% of cases, respectively, are likely to act as two major oncogenic drivers in HCC. This study also identifies several prevalent and potentially actionable mutations, including activating mutations of Janus kinase 1 (JAK1), in 9.1% of patients and provides a path toward therapeutic intervention of the disease.
Figures
Similar articles
-
Hepatitis B virus-associated hepatocellular carcinoma from India: role of viral genotype and mutations in CTNNB1 (beta-catenin) and TP53 genes.J Gastrointest Cancer. 2011 Mar;42(1):20-5. doi: 10.1007/s12029-010-9222-4. J Gastrointest Cancer. 2011. PMID: 20963515 Free PMC article.
-
Integrative analysis of aberrant Wnt signaling in hepatitis B virus-related hepatocellular carcinoma.World J Gastroenterol. 2015 May 28;21(20):6317-28. doi: 10.3748/wjg.v21.i20.6317. World J Gastroenterol. 2015. PMID: 26034368 Free PMC article. Review.
-
Mutations in TP53, CTNNB1 and PIK3CA genes in hepatocellular carcinoma associated with hepatitis B and hepatitis C virus infections.Genomics. 2013 Aug;102(2):74-83. doi: 10.1016/j.ygeno.2013.04.001. Epub 2013 Apr 11. Genomics. 2013. PMID: 23583669 Review.
-
Comprehensive analyses of mutations and hepatitis B virus integration in hepatocellular carcinoma with clinicopathological features.J Gastroenterol. 2016 May;51(5):473-86. doi: 10.1007/s00535-015-1126-4. Epub 2015 Nov 9. J Gastroenterol. 2016. PMID: 26553052
-
Whole-genome sequencing reveals the evolutionary trajectory of HBV-related hepatocellular carcinoma early recurrence.Signal Transduct Target Ther. 2022 Jan 26;7(1):24. doi: 10.1038/s41392-021-00838-3. Signal Transduct Target Ther. 2022. PMID: 35078970 Free PMC article.
Cited by
-
Inside Perspective of the Synthetic and Computational Toolbox of JAK Inhibitors: Recent Updates.Molecules. 2020 Jul 22;25(15):3321. doi: 10.3390/molecules25153321. Molecules. 2020. PMID: 32707925 Free PMC article. Review.
-
Mutation Detection in an Antibody-Producing Chinese Hamster Ovary Cell Line by Targeted RNA Sequencing.Biomed Res Int. 2016;2016:8356435. doi: 10.1155/2016/8356435. Epub 2016 Mar 20. Biomed Res Int. 2016. PMID: 27088091 Free PMC article.
-
Identification and validation of three core genes in p53 signaling pathway in hepatitis B virus-related hepatocellular carcinoma.World J Surg Oncol. 2021 Mar 8;19(1):66. doi: 10.1186/s12957-021-02174-w. World J Surg Oncol. 2021. PMID: 33685467 Free PMC article.
-
Genomic Landscape of HCC.Curr Hepatol Rep. 2020 Dec;19(4):448-461. doi: 10.1007/s11901-020-00553-7. Epub 2020 Nov 10. Curr Hepatol Rep. 2020. PMID: 33816052 Free PMC article.
-
Genomic modeling of hepatitis B virus integration frequency in the human genome.PLoS One. 2019 Jul 29;14(7):e0220376. doi: 10.1371/journal.pone.0220376. eCollection 2019. PLoS One. 2019. PMID: 31356634 Free PMC article.
References
-
- Aguilar F, Harris CC, Sun T, Hollstein M, Cerutti P 1994. Geographic variation of p53 mutational profile in nonmalignant human liver. Science 264: 1317–1319 - PubMed
-
- Audeh MW, Carmichael J, Penson RT, Friedlander M, Powell B, Bell-McGuinn KM, Scott C, Weitzel JN, Oaknin A, Loman N, et al. 2010. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: A proof-of-concept trial. Lancet 376: 245–251 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous