

Characterization of three vasopressin receptor 2 variants: an apparent polymorphism (V266A) and two loss-of-function mutations (R181C and M311V)
- PMID: 23762448
- PMCID: PMC3675069
- DOI: 10.1371/journal.pone.0065885
Characterization of three vasopressin receptor 2 variants: an apparent polymorphism (V266A) and two loss-of-function mutations (R181C and M311V)
Abstract
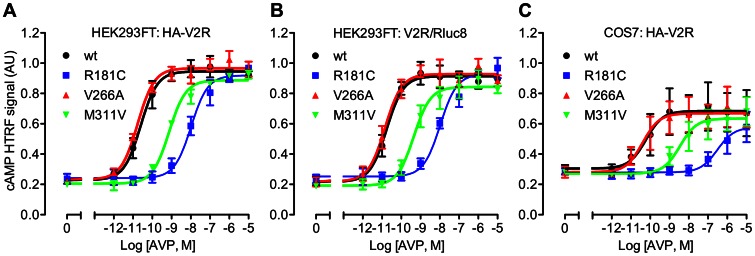
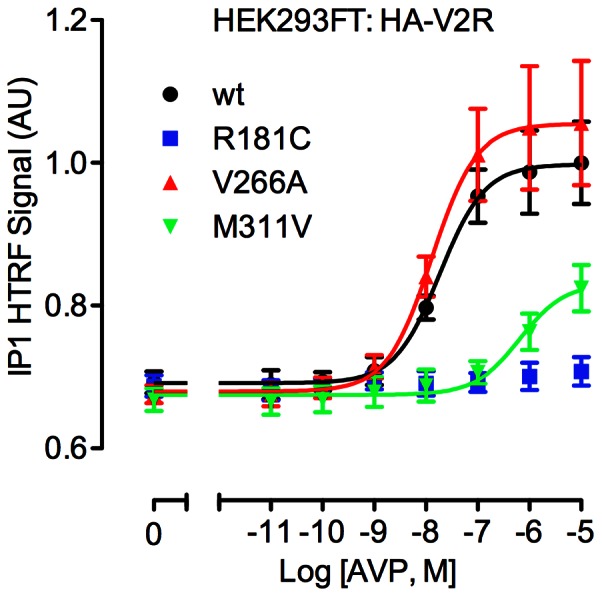
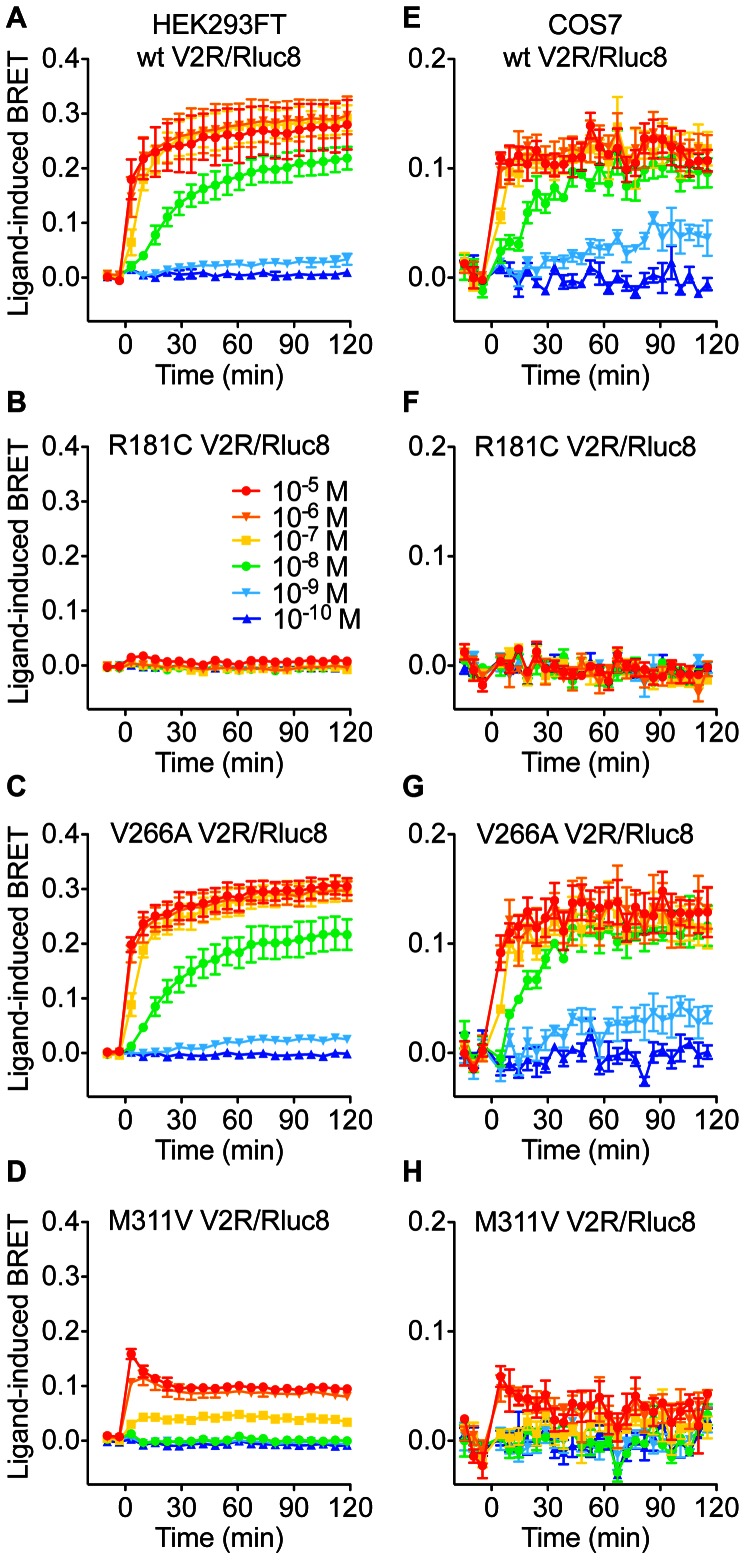
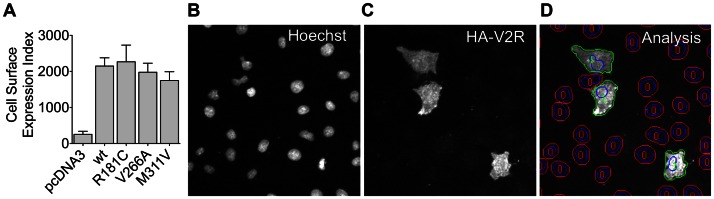
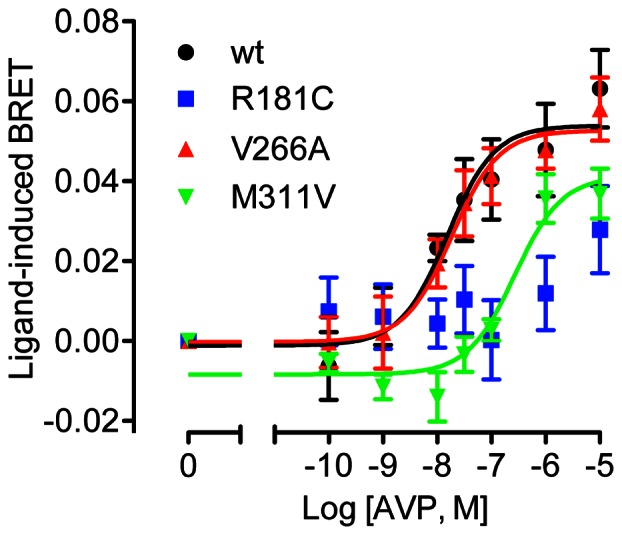
Arginine vasopressin (AVP) is released from the posterior pituitary and controls water homeostasis. AVP binding to vasopressin V2 receptors (V2Rs) located on kidney collecting duct epithelial cells triggers activation of Gs proteins, leading to increased cAMP levels, trafficking of aquaporin-2 water channels, and consequent increased water permeability and antidiuresis. Typically, loss-of-function V2R mutations cause nephrogenic diabetes insipidus (NDI), whereas gain-of-function mutations cause nephrogenic syndrome of inappropriate antidiuresis (NSIAD). Here we provide further characterization of two mutant V2Rs, R181C and M311V, reported to cause complete and partial NDI respectively, together with a V266A variant, in a patient diagnosed with NSIAD. Our data in HEK293FT cells revealed that for cAMP accumulation, AVP was about 500- or 30-fold less potent at the R181C and M311V mutants than at the wild-type receptor respectively (and about 4000- and 60-fold in COS7 cells respectively). However, in contrast to wild type V2R, the R181C mutant failed to increase inositol phosphate production, while with the M311V mutant, AVP exhibited only partial agonism in addition to a 37-fold potency decrease. Similar responses were detected in a BRET assay for β-arrestin recruitment, with the R181C receptor unresponsive to AVP, and partial agonism with a 23-fold decrease in potency observed with M311V in both HEK293FT and COS7 cells. Notably, the V266A V2R appeared functionally identical to the wild-type receptor in all assays tested, including cAMP and inositol phosphate accumulation, β-arrestin interaction, and in a BRET assay of receptor ubiquitination. Each receptor was expressed at comparable levels. Hence, the M311V V2R retains greater activity than the R181C mutant, consistent with the milder phenotype of NDI associated with this mutant. Notably, the R181C mutant appears to be a Gs protein-biased receptor incapable of signaling to inositol phosphate or recruiting β-arrestin. The etiology of NSIAD in the patient with V266A V2R remains unknown.
Conflict of interest statement
Figures

Similar articles
-
Functional characterization of vasopressin type 2 receptor substitutions (R137H/C/L) leading to nephrogenic diabetes insipidus and nephrogenic syndrome of inappropriate antidiuresis: implications for treatments.Mol Pharmacol. 2010 May;77(5):836-45. doi: 10.1124/mol.109.061804. Epub 2010 Feb 16. Mol Pharmacol. 2010. PMID: 20159941 Free PMC article.
-
Mutations of Vasopressin Receptor 2 Including Novel L312S Have Differential Effects on Trafficking.Mol Endocrinol. 2016 Aug;30(8):889-904. doi: 10.1210/me.2016-1002. Epub 2016 Jun 29. Mol Endocrinol. 2016. PMID: 27355191 Free PMC article.
-
Gain-of-function mutations of the V2 vasopressin receptor in nephrogenic syndrome of inappropriate antidiuresis (NSIAD): a cell-based assay to assess constitutive water reabsorption.Pflugers Arch. 2019 Oct;471(10):1291-1304. doi: 10.1007/s00424-019-02307-x. Epub 2019 Sep 5. Pflugers Arch. 2019. PMID: 31486901
-
[Vasopressin V2 receptor-related pathologies: congenital nephrogenic diabetes insipidus and nephrogenic syndrome of inappropiate antidiuresis].Nephrol Ther. 2014 Dec;10(7):538-46. doi: 10.1016/j.nephro.2014.09.002. Epub 2014 Oct 25. Nephrol Ther. 2014. PMID: 25449762 Review. French.
-
[Nephrogenic syndrome of inappropriate antidiuresis].Pan Afr Med J. 2019 Apr 29;32:210. doi: 10.11604/pamj.2019.32.210.6006. eCollection 2019. Pan Afr Med J. 2019. PMID: 31312322 Free PMC article. Review. French.
Cited by
-
A structural basis for how ligand binding site changes can allosterically regulate GPCR signaling and engender functional selectivity.Sci Signal. 2020 Feb 4;13(617):eaaw5885. doi: 10.1126/scisignal.aaw5885. Sci Signal. 2020. PMID: 32019899 Free PMC article.
-
Vasopressin regulates the growth of the biliary epithelium in polycystic liver disease.Lab Invest. 2016 Nov;96(11):1147-1155. doi: 10.1038/labinvest.2016.93. Epub 2016 Aug 29. Lab Invest. 2016. PMID: 27571215 Free PMC article.
-
Biased signaling in naturally occurring mutations of G protein-coupled receptors associated with diverse human diseases.Biochim Biophys Acta Mol Basis Dis. 2021 Jan 1;1867(1):165973. doi: 10.1016/j.bbadis.2020.165973. Epub 2020 Sep 17. Biochim Biophys Acta Mol Basis Dis. 2021. PMID: 32949766 Free PMC article. Review.
-
Signaling Modification by GPCR Heteromer and Its Implication on X-Linked Nephrogenic Diabetes Insipidus.PLoS One. 2016 Sep 20;11(9):e0163086. doi: 10.1371/journal.pone.0163086. eCollection 2016. PLoS One. 2016. PMID: 27649563 Free PMC article.
-
Agonist-induced formation of unproductive receptor-G12 complexes.Proc Natl Acad Sci U S A. 2020 Sep 1;117(35):21723-21730. doi: 10.1073/pnas.2003787117. Epub 2020 Aug 17. Proc Natl Acad Sci U S A. 2020. PMID: 32817560 Free PMC article.
References
-
- Ball SG (2007) Vasopressin and disorders of water balance: the physiology and pathophysiology of vasopressin. Ann Clin Biochem 44: 417–431. - PubMed
-
- Morello J-P, Bichet DG (2001) Nephrogenic Diabetes Insipidus. Annu Rev Physiol 63: 607–630. - PubMed
-
- Sands JM, Bichet DG (2006) Nephrogenic Diabetes Insipidus. Ann Intern Med 144: 186–194. - PubMed
-
- Lolait SJ, O'Carroll A-M, McBride OW, Konig M, Morel A, et al. (1992) Cloning and characterization of a vasopressin V2 receptor and possible link to nephrogenic diabetes insipidus. Nature 357: 336–339. - PubMed
-
- Birnbaumer M, Seibold A, Gilbert S, Ishido M, Barberis C, et al. (1992) Molecular cloning of the receptor for human antidiuretic hormone. Nature 357: 333–335. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
