

Nrf2 deficiency prevents reductive stress-induced hypertrophic cardiomyopathy
- PMID: 23761402
- PMCID: PMC3778956
- DOI: 10.1093/cvr/cvt150
Nrf2 deficiency prevents reductive stress-induced hypertrophic cardiomyopathy
Abstract
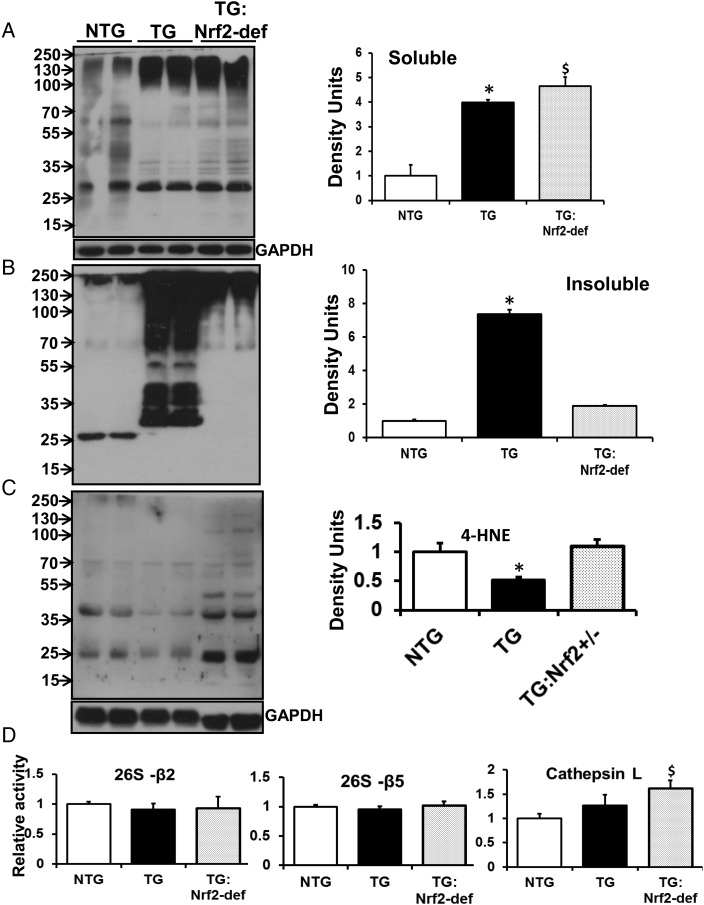
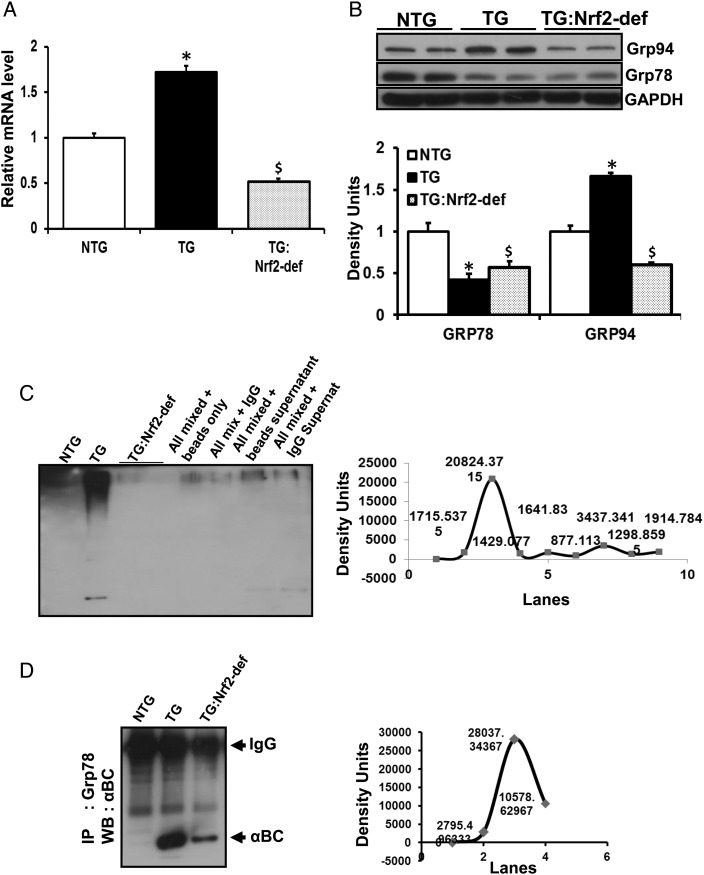
Aims: Mutant protein aggregation (PA) cardiomyopathy (MPAC) is characterized by reductive stress (RS), PA (of chaperones and cytoskeletal components), and ventricular dysfunction in transgenic mice expressing human mutant CryAB (hmCryAB). Sustained activation of nuclear erythroid-2 like factor-2 (Nrf2) causes RS, which contributes to proteotoxic cardiac disease. The goals of this pre-clinical study were to (i) investigate whether disrupting Nrf2-antioxidant signalling prevents RS and rescues redox homeostasis in hearts expressing the mutant chaperone and (ii) elucidate mechanisms that could delay proteotoxic cardiac disease.
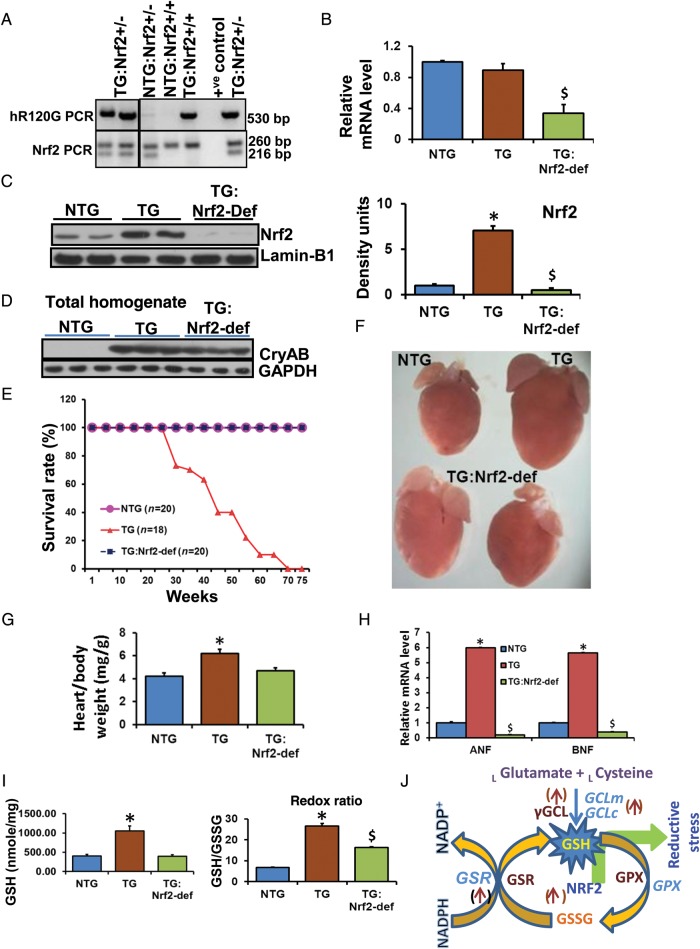
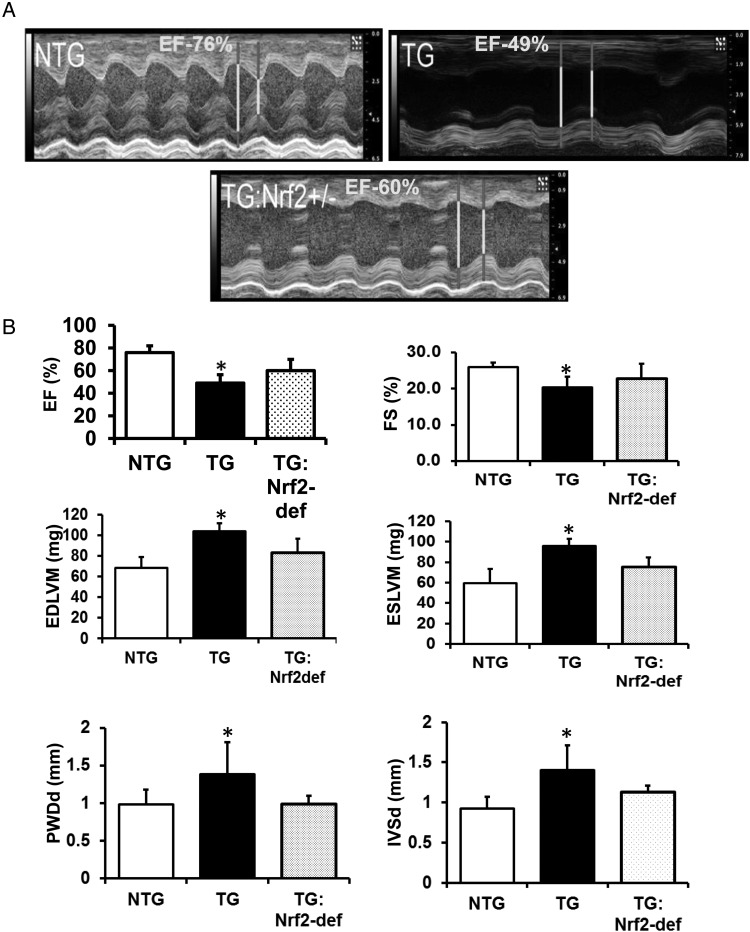
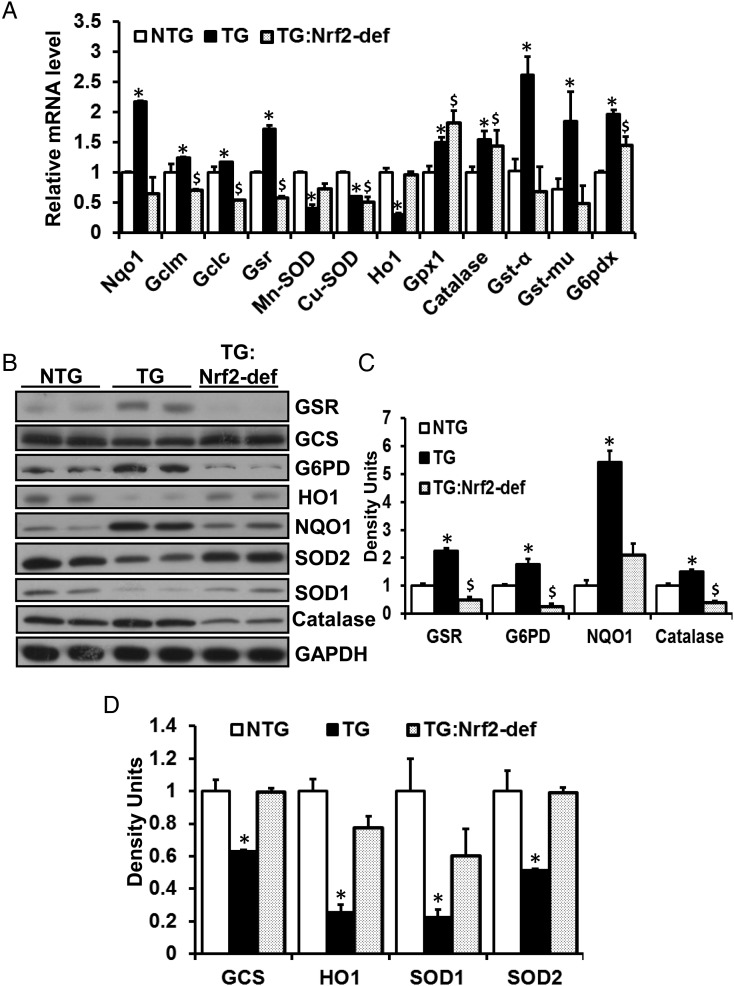
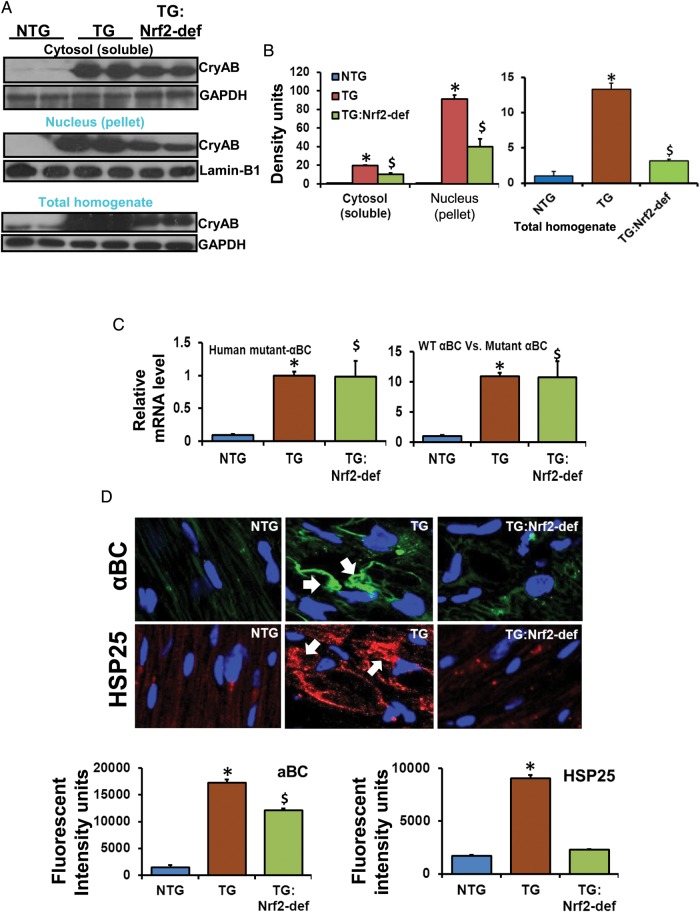
Methods and results: Non-transgenic (NTG), transgenic (TG) with MPAC and MPAC-TG:Nrf2-deficient (Nrf2-def) mice were used in this study. The effects of Nrf2 diminution (Nrf2±) on RS mediated MPAC in TG mice were assessed at 6-7 and 10 months of age. The diminution of Nrf2 prevented RS and prolonged the survival of TG mice (∼50 weeks) by an additional 20-25 weeks. The TG:Nrf2-def mice did not exhibit cardiac hypertrophy at even 60 weeks, while the MPAC-TG mice developed pathological hypertrophy and heart failure starting at 24-28 weeks of age. Aggregation of cardiac proteins was significantly reduced in TG:Nrf2-def when compared with TG mice at 7 months. Preventing RS and maintaining redox homeostasis in the TG:Nrf2-def mice ameliorated PA, leading to decreased ubiquitination of proteins.
Conclusion: Nrf2 deficiency rescues redox homeostasis, which reduces aggregation of mutant proteins, thereby delaying the proteotoxic pathological cardiac remodelling caused by RS and toxic protein aggregates.
Keywords: Nrf2; antioxidants; cardiomyopathy; protein aggregation; reductive stress.
Figures
Comment in
-
Reducing damage through Nrf-2.Cardiovasc Res. 2013 Oct 1;100(1):1-3. doi: 10.1093/cvr/cvt204. Epub 2013 Aug 22. Cardiovasc Res. 2013. PMID: 23970486 No abstract available.
Similar articles
-
Reductive Stress Causes Pathological Cardiac Remodeling and Diastolic Dysfunction.Antioxid Redox Signal. 2020 Jun;32(18):1293-1312. doi: 10.1089/ars.2019.7808. Antioxid Redox Signal. 2020. PMID: 32064894 Free PMC article.
-
Transgenic Expression of Nrf2 Induces a Pro-Reductive Stress and Adaptive Cardiac Remodeling in the Mouse.Genes (Basel). 2022 Aug 24;13(9):1514. doi: 10.3390/genes13091514. Genes (Basel). 2022. PMID: 36140682 Free PMC article.
-
Sustained activation of nuclear erythroid 2-related factor 2/antioxidant response element signaling promotes reductive stress in the human mutant protein aggregation cardiomyopathy in mice.Antioxid Redox Signal. 2011 Mar 15;14(6):957-71. doi: 10.1089/ars.2010.3587. Epub 2011 Feb 2. Antioxid Redox Signal. 2011. PMID: 21126175 Free PMC article.
-
Tandem Mass Tagging Based Identification of Proteome Signatures for Reductive Stress Cardiomyopathy.Front Cardiovasc Med. 2022 Jun 13;9:848045. doi: 10.3389/fcvm.2022.848045. eCollection 2022. Front Cardiovasc Med. 2022. PMID: 35770227 Free PMC article.
-
Reductive potential - a savior turns stressor in protein aggregation cardiomyopathy.Biochim Biophys Acta. 2015 Jan;1852(1):53-60. doi: 10.1016/j.bbadis.2014.11.010. Epub 2014 Nov 15. Biochim Biophys Acta. 2015. PMID: 25446995 Free PMC article. Review.
Cited by
-
Dimethyl Fumarate Prevents the Development of Chronic Social Stress-Induced Hypertension in Borderline Hypertensive Rats.Antioxidants (Basel). 2024 Aug 3;13(8):947. doi: 10.3390/antiox13080947. Antioxidants (Basel). 2024. PMID: 39199192 Free PMC article.
-
Exploratory bibliometric analysis and text mining to reveal research trends in cardiac aging.Aging Med (Milton). 2024 Jun 14;7(3):301-311. doi: 10.1002/agm2.12329. eCollection 2024 Jun. Aging Med (Milton). 2024. PMID: 38975309 Free PMC article.
-
Sleep fragmentation induces heart failure in a hypertrophic cardiomyopathy mouse model by altering redox metabolism.iScience. 2024 Feb 1;27(3):109075. doi: 10.1016/j.isci.2024.109075. eCollection 2024 Mar 15. iScience. 2024. PMID: 38361607 Free PMC article.
-
Thioredoxin (Trx): A redox target and modulator of cellular senescence and aging-related diseases.Redox Biol. 2024 Apr;70:103032. doi: 10.1016/j.redox.2024.103032. Epub 2024 Jan 13. Redox Biol. 2024. PMID: 38232457 Free PMC article. Review.
-
Interplay between Nrf2 and αB-crystallin in the lens and heart of zebrafish under proteostatic stress.Front Mol Biosci. 2023 Jul 28;10:1185704. doi: 10.3389/fmolb.2023.1185704. eCollection 2023. Front Mol Biosci. 2023. PMID: 37577747 Free PMC article.
References
-
- Lloyd-Jones D, Adams R, Carnethon M, De Simone G, Ferguson TB, Flegal K, et al. Heart disease and stroke statistics—2009 update: a report from the American Heart Association statistics committee and stroke statistics subcommittee. Circulation. 2009;119:480–486. doi: 10.1161/CIRCULATIONAHA.108.191259. - DOI - PubMed
-
- Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB, et al. American Heart Association Statistics C, Stroke Statistics S. Heart disease and stroke statistics—2012 update: a report from the American Heart Association. Circulation. 2012;125:e2–e220. doi: 10.1161/CIR.0b013e31823ac046. - DOI - PMC - PubMed
-
- Maron BJ, Gardin JM, Flack JM, Gidding SS, Kurosaki TT, Bild DE. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the cardia study. Coronary artery risk development in (young) adults. Circulation. 1995;92:785–789. doi: 10.1161/01.CIR.92.4.785. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
