

Review
doi: 10.1038/nri3452.
Activation and regulation of the inflammasomes
Affiliations
- PMID: 23702978
- PMCID: PMC3807999
- DOI: 10.1038/nri3452
Item in Clipboard
Review
Activation and regulation of the inflammasomes
Nat Rev Immunol.
2013 Jun.
Abstract
Inflammasomes are key signalling platforms that detect pathogenic microorganisms and sterile stressors, and that activate the highly pro-inflammatory cytokines interleukin-1β (IL-1β) and IL-18. In this Review, we discuss the complex regulatory mechanisms that facilitate a balanced but effective inflammasome-mediated immune response, and we highlight the similarities to another molecular signalling platform - the apoptosome - that monitors cellular health. Extracellular regulatory mechanisms are discussed, as well as the intracellular control of inflammasome assembly, for example, via ion fluxes, free radicals and autophagy.
Figures
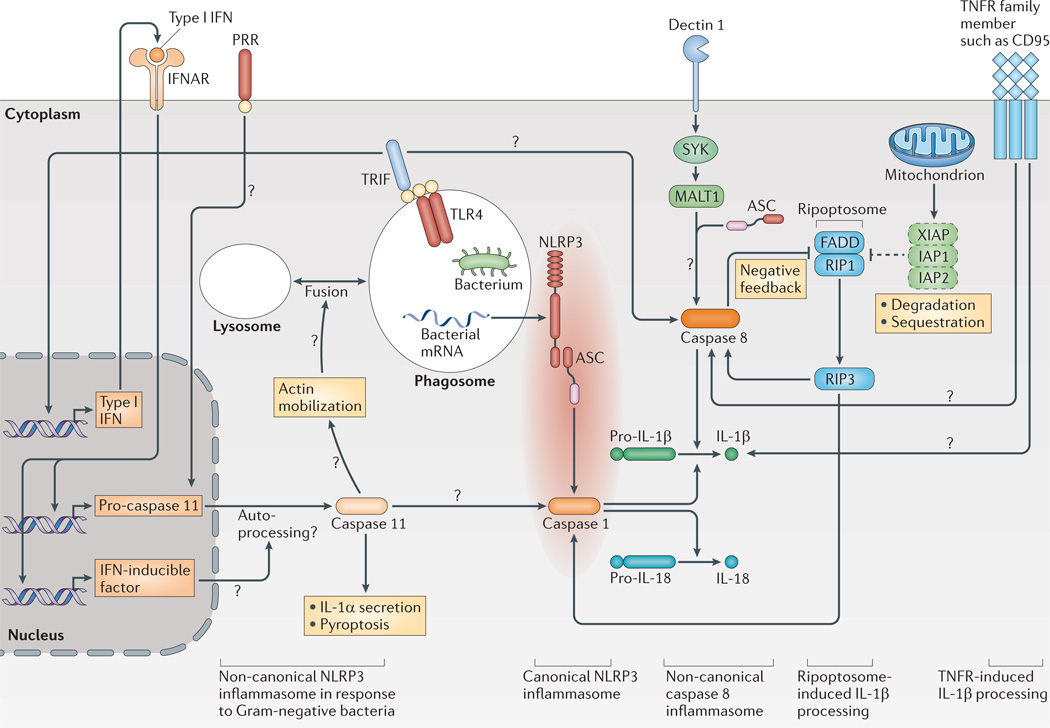
NLRP3 (NOD-, LRR- and pyrin domain-containing 3) needs additional cofactors for the processing of interleukin-1β (IL-1β) in response to Gram-negative bacteria; this path way has been termed the non-canonical NLRP3 inflammasome. Toll-like receptor 4 (TLR4) signalling via TIR domain-containing adaptor protein inducing IFNβ (TRIF) induces the secretion of type I interferons (IFNs), which lead to the activation of caspase 11 via autocrine signalling through the IFNα/β receptor (IFNAR), possibly involving additional (not yet known) IFN-inducible factors. Caspase 11 is necessary for the activation of caspase 1 and has an independent role in IL-1α secretion and pyroptosis in response to Gram-negative bacteria. A possible mechanism for caspase 11 action might be its role in actin mobilization via cofilin, which might lead to increased fusion of lysosomes to phagosomes and, potentially, to increased leakage of bacterial mRNA (also termed vita-PAMPs) to the cytoplasm to activate NLRP3 (not shown). A platform consisting of mucosa-associated lymphoid tissue lymphoma translocation protein 1 (MALT1), caspase 8 and the adaptor protein ASC (termed the non-canonical caspase 8 inflammasome) is formed in response to stimulation of dectin 1. Caspase 8 might also be activated by TLRs using the signalling adaptor TRIF in the presence of cycloheximide. In addition, the formation of the ripoptosome is triggered by the loss of inhibitor of apoptosis proteins (IAPs) with concurrent TLR stimulation. The ripoptosome — consisting of FAS-associated death domain protein (FADD) and receptor-interacting protein 1 (RIP1) — activates caspase 8 via RIP3. However, caspase 8 can also limit ripoptosome action on NLRP3. Members of the tumour necrosis factor receptor (TNFR) family might also induce pro-IL-1β cleavage, as has been shown for CD95 (also known as FAS). Question marks show pathways that are still speculative. PRR, pattern recognition receptor; XIAP, X-linked inhibitor of apoptosis protein.
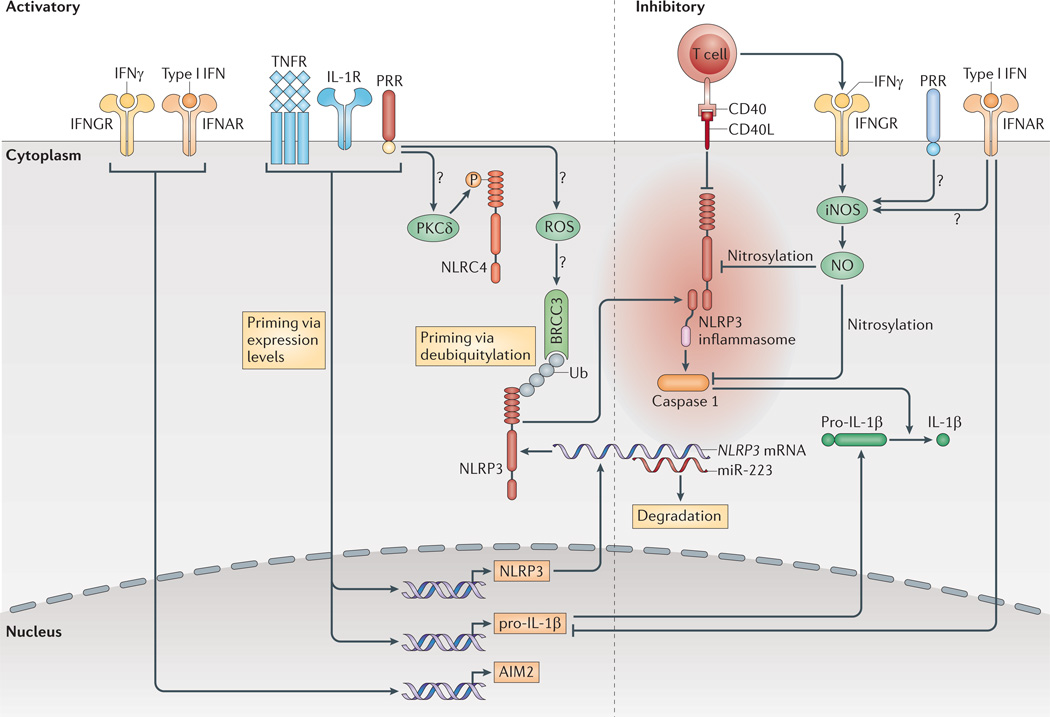
Pro-interleukin-1β (pro-IL-1β) and NLRP3 (NOD-, LRR-and pyrin domain-containing 3) expression are induced by transcriptionally active pattern recognition receptors (PRRs) or by cytokine receptors. Furthermore, NLRP3 deubiquitylation by the K63-specific deubiquitinase BRCC3 is c rucial for its activation. Direct contact with mature or memory T cells inhibits the inflammasomes, probably via tumour necrosis factor receptor (TNFR) superfamily interactions. Type I interferons (IFNs) inhibit the transcription of pro-IL-1β , but also upregulate the expression of absent in melanoma 2 (AIM2). Both type I IFNs and IFNγ inhibit NLRP3 through the induction of nitric oxide (NO) via inducible nitric oxide synthase (iNOS), possibly with the requirement of concomitant priming by PRRs. Ub shows ubiquitylated proteins. Question marks show pathways that are still speculative. CD40L, CD40 ligand; IFNAR, interferon-α/β receptor; IFNGR, interferon-γ receptor; IL-1R, IL-1 receptor; miR-223, microRNA-223; NLRC4, NOD-, LRR- and CARD-containing 4; ROS, reactive oxygen species; PKCδ, protein kinase Cδ.
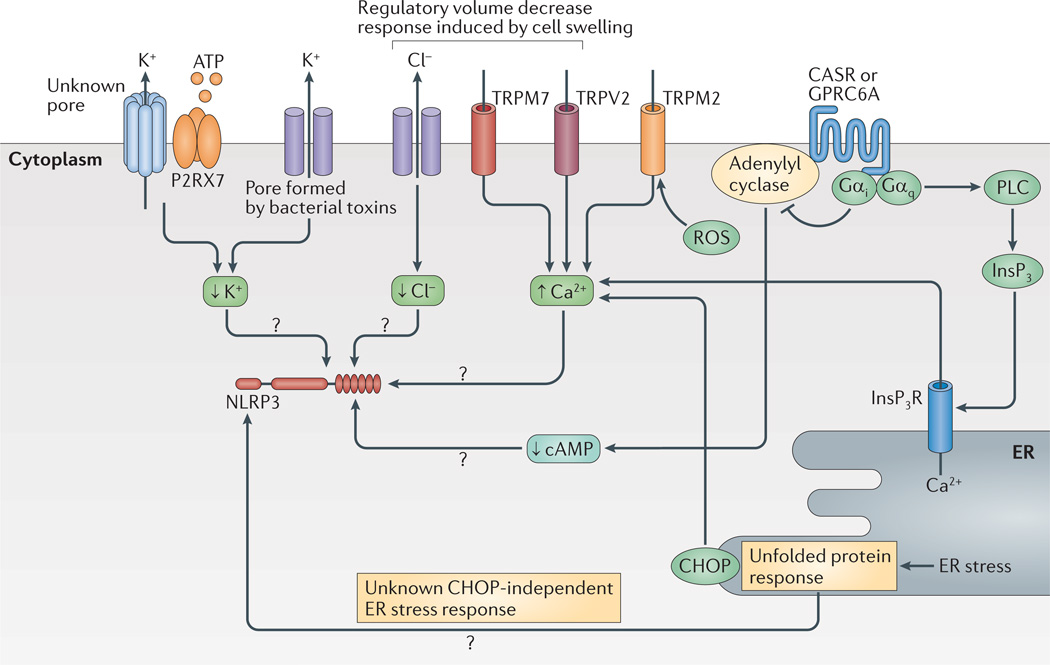
K+ and Cl− efflux, as well as Ca2+ mobilization, have a role in the regulation of NLRP3 (NOD-, LRR- and pyrin domain-containing 3). K+ efflux is either achieved directly by pore-forming toxins such as nigericin or indirectly — for example, via the purinergic receptor for ATP P2RX7. Cl− and Ca2+ ion fluxes can be regulated during the regulatory volume decrease response by transient receptor potential receptors (TRPV2 or TRPM7). In addition, reactive oxygen species (ROS) can activate TRPM2 for Ca2+ influx. Ca2+ is also regulated by the unfolded protein response via C/EPB-homologous protein (CHOP). The unfolded protein response is also implicated in other pathways that regulate NLRP3. G-protein coupled receptors (GPCRs) such as calcium-sensing receptor (CASR) and GPRC6A regulate both Ca2+ levels and cyclic AMP (cAMP) levels via activation of phospholipase C (PLC) or inhibition of adenylyl cyclase, respectively. High cAMP levels might directly inhibit NLRP3 (not shown). Inositol-1,4,5-trisphosphate (InsP3) that is generated by PLC leads to release of Ca2+ from the endoplasmic reticulum (ER). How Ca2+ influences NLRP3 activation is not fully understood but it has many molecular targets. Question marks show pathways that are still speculative. InsP3R, InsP3 receptor.
Similar articles
-
IL-1β and IL-18: inflammatory markers or mediators of hypertension?Br J Pharmacol. 2014 Dec;171(24):5589-602. doi: 10.1111/bph.12876. Br J Pharmacol. 2014. PMID: 25117218 Free PMC article. Review.
-
The NLRP3 inflammasome in kidney disease and autoimmunity.Nephrology (Carlton). 2016 Sep;21(9):736-44. doi: 10.1111/nep.12785. Nephrology (Carlton). 2016. PMID: 27011059 Review.
-
The Nlrp3 inflammasome promotes myocardial dysfunction in structural cardiomyopathy through interleukin-1β.Exp Physiol. 2013 Feb;98(2):462-72. doi: 10.1113/expphysiol.2012.068338. Epub 2012 Jul 30. Exp Physiol. 2013. PMID: 22848083
-
Methylsulfonylmethane inhibits NLRP3 inflammasome activation.Cytokine. 2015 Feb;71(2):223-31. doi: 10.1016/j.cyto.2014.11.001. Epub 2014 Nov 21. Cytokine. 2015. PMID: 25461402
-
Cutting edge: the NLRP3 inflammasome links complement-mediated inflammation and IL-1β release.J Immunol. 2013 Aug 1;191(3):1006-10. doi: 10.4049/jimmunol.1300489. Epub 2013 Jul 1. J Immunol. 2013. PMID: 23817414 Free PMC article.
Cited by
-
Activators of SIRT1 in the kidney and protective effects of SIRT1 during acute kidney injury (AKI) (effect of SIRT1 activators on acute kidney injury).Clin Exp Nephrol. 2021 Aug;25(8):807-821. doi: 10.1007/s10157-021-02057-0. Epub 2021 Mar 29. Clin Exp Nephrol. 2021. PMID: 33779856 Review.
-
NLRP3 Inflammasome/Pyroptosis: A Key Driving Force in Diabetic Cardiomyopathy.Int J Mol Sci. 2022 Sep 13;23(18):10632. doi: 10.3390/ijms231810632. Int J Mol Sci. 2022. PMID: 36142531 Free PMC article. Review.
-
A proteomics perspective on viral DNA sensors in host defense and viral immune evasion mechanisms.J Mol Biol. 2015 Jun 5;427(11):1995-2012. doi: 10.1016/j.jmb.2015.02.016. Epub 2015 Feb 26. J Mol Biol. 2015. PMID: 25728651 Free PMC article. Review.
-
The phagosomal solute transporter SLC15A4 promotes inflammasome activity via mTORC1 signaling and autophagy restraint in dendritic cells.EMBO J. 2022 Oct 17;41(20):e111161. doi: 10.15252/embj.2022111161. Epub 2022 Aug 29. EMBO J. 2022. PMID: 36031853 Free PMC article.
-
Lung Ischemia-Reperfusion is a Sterile Inflammatory Process Influenced by Commensal Microbiota in Mice.Shock. 2015 Sep;44(3):272-9. doi: 10.1097/SHK.0000000000000415. Shock. 2015. PMID: 26196836 Free PMC article.
References
-
- Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34:637–650. - PubMed
-
- Kato H, Takahasi K, Fujita T. RIG-I-like receptors: cytoplasmic sensors for non-self RNA. Immunol. Rev. 2011;243:91–98. - PubMed
-
- Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol. Cell. 2002;10:417–426. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
