

Prediction of clustered RNA-binding protein motif sites in the mammalian genome
- PMID: 23685613
- PMCID: PMC3737533
- DOI: 10.1093/nar/gkt421
Prediction of clustered RNA-binding protein motif sites in the mammalian genome
Abstract
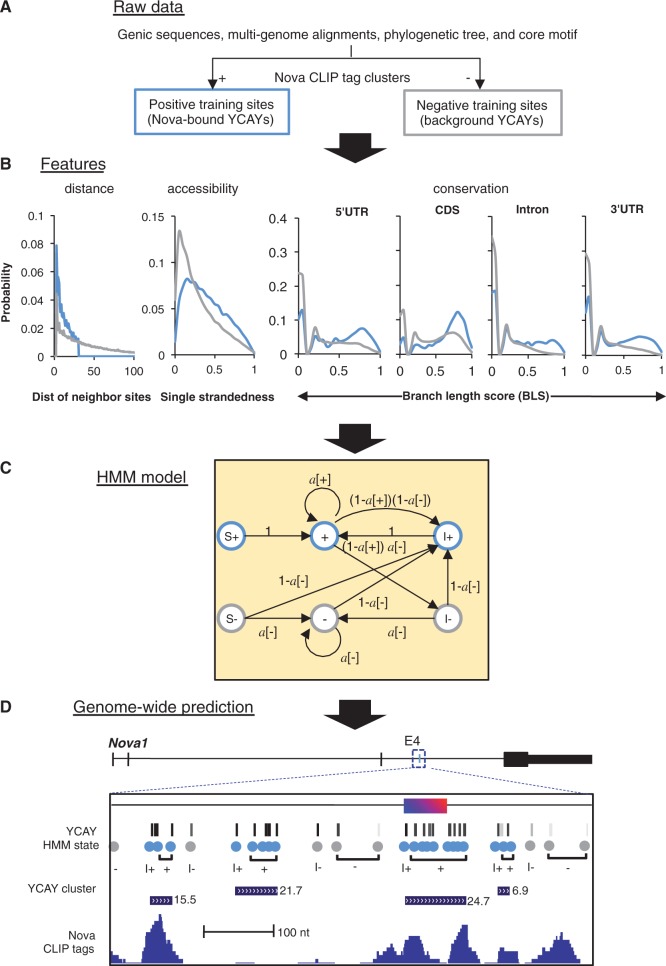
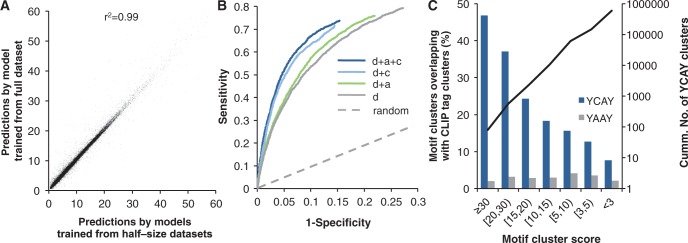
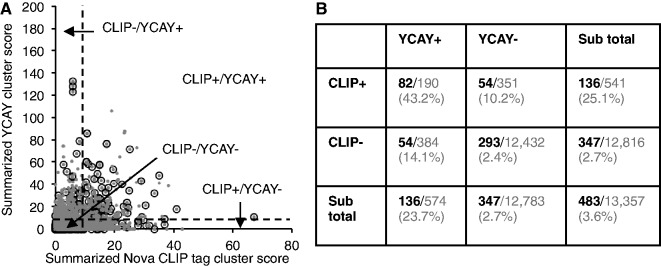
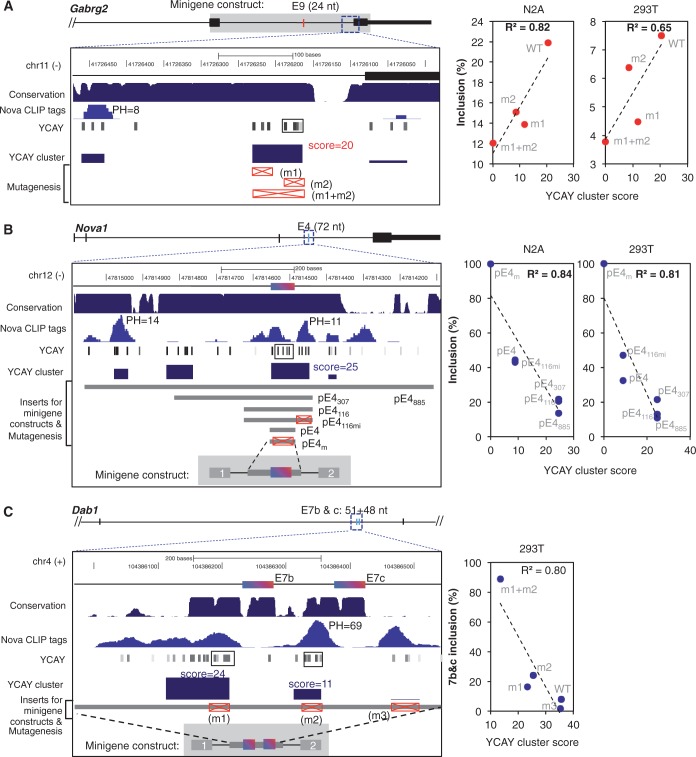
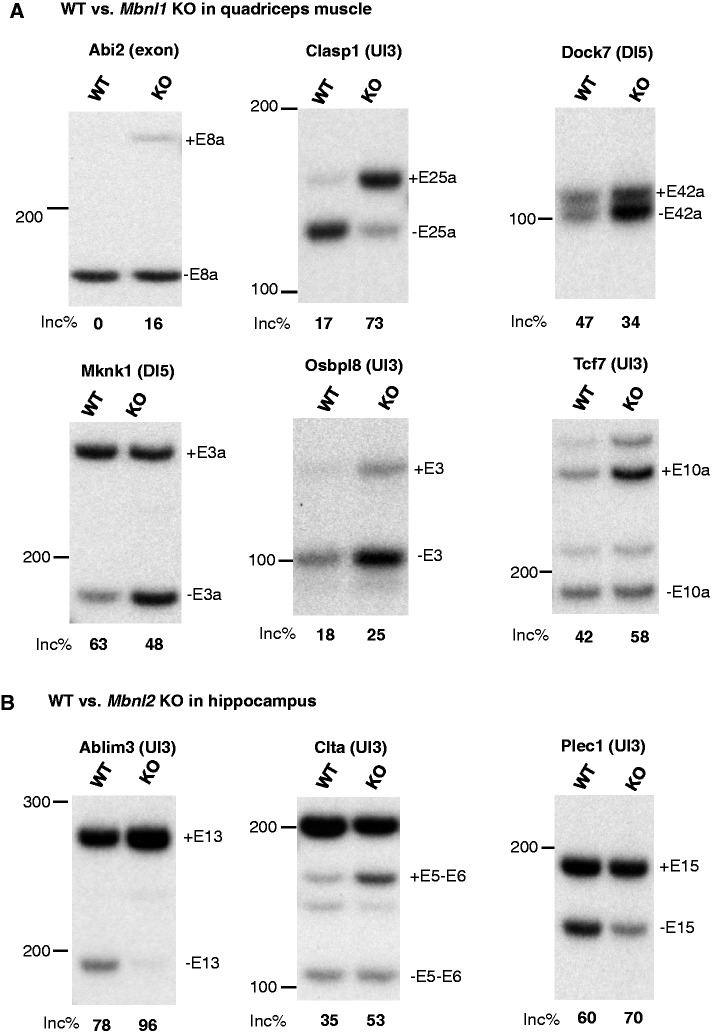
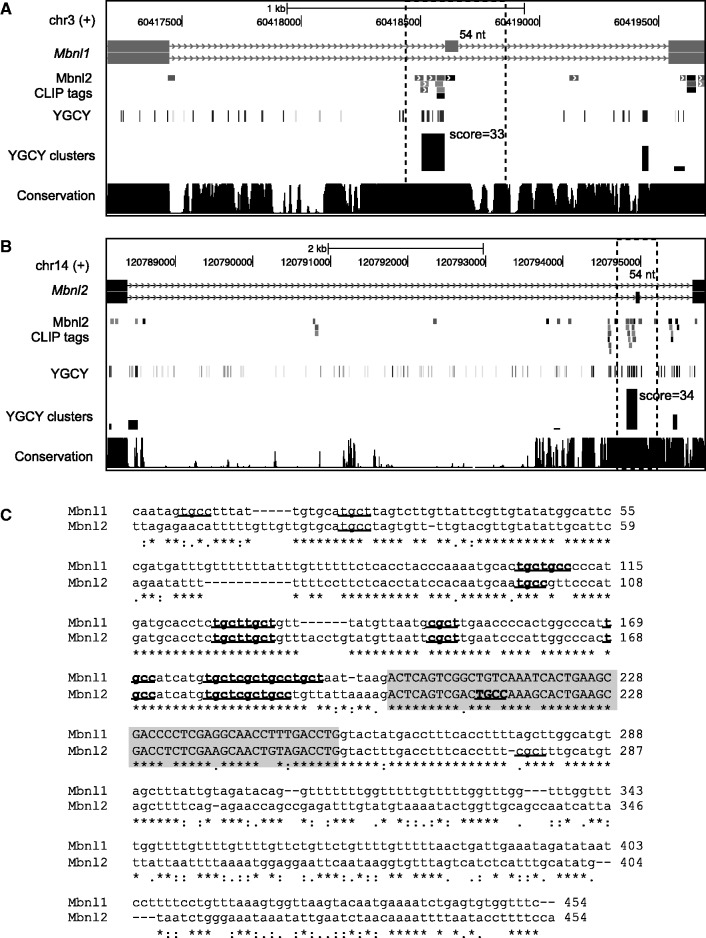
Sequence-specific interactions of RNA-binding proteins (RBPs) with their target transcripts are essential for post-transcriptional gene expression regulation in mammals. However, accurate prediction of RBP motif sites has been difficult because many RBPs recognize short and degenerate sequences. Here we describe a hidden Markov model (HMM)-based algorithm mCarts to predict clustered functional RBP-binding sites by effectively integrating the number and spacing of individual motif sites, their accessibility in local RNA secondary structures and cross-species conservation. This algorithm learns and quantifies rules of these features, taking advantage of a large number of in vivo RBP-binding sites obtained from cross-linking and immunoprecipitation data. We applied this algorithm to study two representative RBP families, Nova and Mbnl, which regulate tissue-specific alternative splicing through interacting with clustered YCAY and YGCY elements, respectively, and predicted their binding sites in the mouse transcriptome. Despite the low information content in individual motif elements, our algorithm made specific predictions for successful experimental validation. Analysis of predicted sites also revealed cases of extensive and distal RBP-binding sites important for splicing regulation. This algorithm can be readily applied to other RBPs to infer their RNA-regulatory networks. The software is freely available at http://zhanglab.c2b2.columbia.edu/index.php/MCarts.
Figures
Similar articles
-
mCarts: Genome-Wide Prediction of Clustered Sequence Motifs as Binding Sites for RNA-Binding Proteins.Methods Mol Biol. 2016;1421:215-26. doi: 10.1007/978-1-4939-3591-8_17. Methods Mol Biol. 2016. PMID: 26965268
-
rMAPS: RNA map analysis and plotting server for alternative exon regulation.Nucleic Acids Res. 2016 Jul 8;44(W1):W333-8. doi: 10.1093/nar/gkw410. Epub 2016 May 12. Nucleic Acids Res. 2016. PMID: 27174931 Free PMC article.
-
RNA Sequence Context Effects Measured In Vitro Predict In Vivo Protein Binding and Regulation.Mol Cell. 2016 Oct 20;64(2):294-306. doi: 10.1016/j.molcel.2016.08.035. Epub 2016 Oct 6. Mol Cell. 2016. PMID: 27720642 Free PMC article.
-
Developing global insight into RNA regulation.Cold Spring Harb Symp Quant Biol. 2006;71:321-7. doi: 10.1101/sqb.2006.71.002. Cold Spring Harb Symp Quant Biol. 2006. PMID: 17381312 Review.
-
Finding the target sites of RNA-binding proteins.Wiley Interdiscip Rev RNA. 2014 Jan-Feb;5(1):111-30. doi: 10.1002/wrna.1201. Epub 2013 Nov 11. Wiley Interdiscip Rev RNA. 2014. PMID: 24217996 Free PMC article. Review.
Cited by
-
Orthogonal matrix factorization enables integrative analysis of multiple RNA binding proteins.Bioinformatics. 2016 May 15;32(10):1527-35. doi: 10.1093/bioinformatics/btw003. Epub 2016 Jan 18. Bioinformatics. 2016. PMID: 26787667 Free PMC article.
-
A novel method for the identification of conserved structural patterns in RNA: From small scale to high-throughput applications.Nucleic Acids Res. 2016 Oct 14;44(18):8600-8609. doi: 10.1093/nar/gkw750. Epub 2016 Aug 31. Nucleic Acids Res. 2016. PMID: 27580722 Free PMC article.
-
How motif environment influences transcription factor search dynamics: Finding a needle in a haystack.Bioessays. 2016 Jul;38(7):605-12. doi: 10.1002/bies.201600005. Epub 2016 May 19. Bioessays. 2016. PMID: 27192961 Free PMC article. Review.
-
GraphProt: modeling binding preferences of RNA-binding proteins.Genome Biol. 2014 Jan 22;15(1):R17. doi: 10.1186/gb-2014-15-1-r17. Genome Biol. 2014. PMID: 24451197 Free PMC article.
-
SeedVicious: Analysis of microRNA target and near-target sites.PLoS One. 2018 Apr 17;13(4):e0195532. doi: 10.1371/journal.pone.0195532. eCollection 2018. PLoS One. 2018. PMID: 29664927 Free PMC article.
References
-
- Licatalosi DD, Darnell RB. Splicing regulation in neurologic disease. Neuron. 2006;52:93–101. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
