

Review
doi: 10.1016/j.tim.2013.04.002.
Epub 2013 May 17.
The emerging world of the fungal microbiome
Affiliations
- PMID: 23685069
- PMCID: PMC3708484
- DOI: 10.1016/j.tim.2013.04.002
Item in Clipboard
Review
The emerging world of the fungal microbiome
Trends Microbiol.
2013 Jul.
Abstract
The study of the fungal microbiota ('mycobiome') is a new and rapidly emerging field that lags behind our understanding of the bacterial microbiome. Every human has fungi as part of their microbiota, but the total number of fungal cells is orders of magnitude smaller than that of the bacterial microbiota. However, the impact of the mycobiome on human health is significant, especially as a reservoir for blooms of pathogenic microbes when the host is compromised and as a potential cofactor in inflammatory diseases and metabolic disorders.
Copyright © 2013 Elsevier Ltd. All rights reserved.
Figures
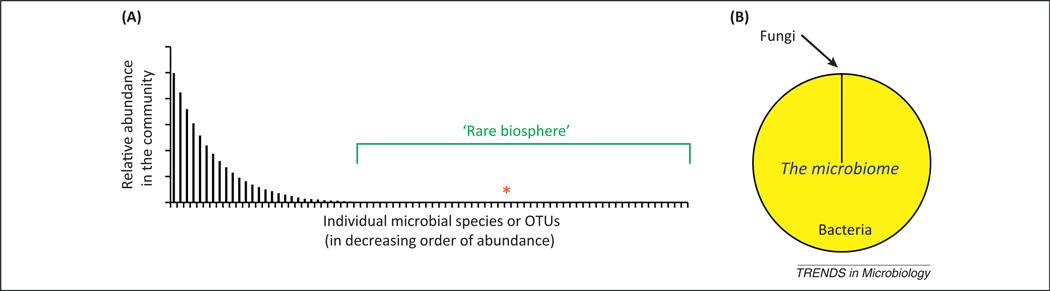
The rare biosphere and the fungal microbiome. (A) Representation of the membership of the microbiome, displayed as a rank abundance curve and illustrating the concept of the ‘rare biosphere’ within the entirety of the microbiome and the relative position of a microbe at 0.01% abundance (asterisk) in this example. In the complex microbial communities of the mucosa, a relatively small number of species dominate, but hundreds to thousands of low abundance species also exist in the communities. At mucosal sites where bacterial abundance can reach >1010/g, a microbe present at 104/g would be <0.0001% of the cellular content of the community. Microbial diversity in the microbiome is extensive and the large number of low abundance species (or operational taxonomic units, OTUs) in mucosal samples have been described as part of a rare biosphere [2,3]. However, the rare biosphere serves as a reservoir for potentially pathogenic microbes, such as members of the fungal microbiome, which grow out (bloom) and cause disease when the mucosal environment is disturbed. The rare biosphere may also harbor species that have a disproportionate effect (positive or negative) on the dominant members of the microbiome, a potential pathway by which the fungal microbiome may promote chronic disease. (B) Estimated relative cellular abundance of fungi to bacteria in human feces, based upon the MetaHIT metagenomic analysis [1].
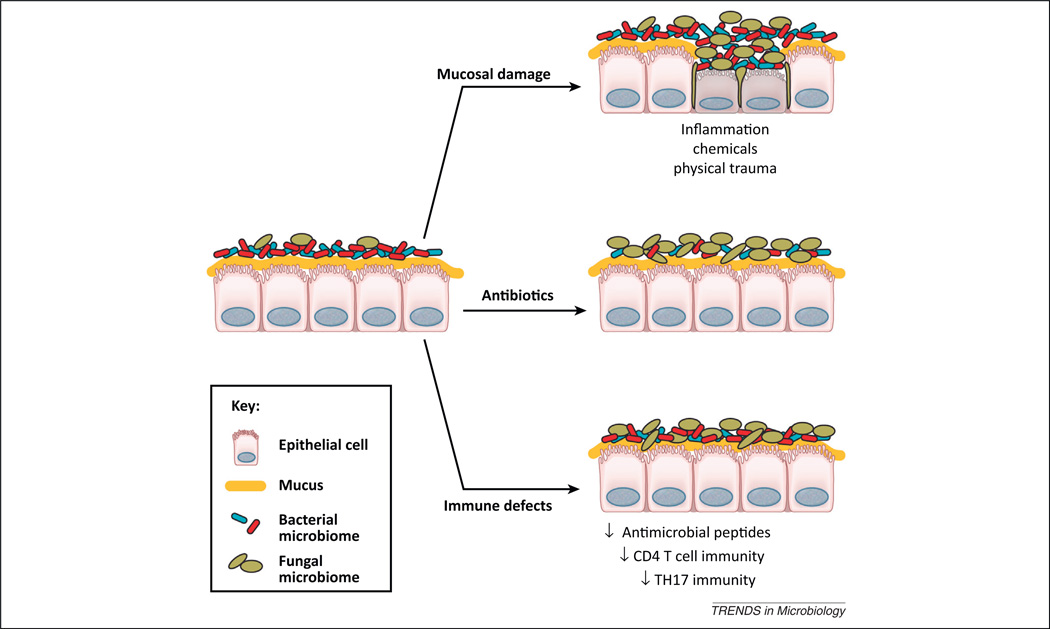
Mechanisms by which the fungal microbiome can grow out and cause disease on mucosal surfaces. The fungal microbiome exists on mucosal surfaces in a symbiotic relationship with the host and the bacterial microbiome. Any of the following will promote the outgrowth of fungi on a mucosal surface: disruption of bacteria-mediated colonization resistance by antibiotics; damage to the mucosa by uncontrolled inflammatory responses, physical damage, or chemical-mediated injury; or defects in host defenses/immunity that affect innate immunity or specific facets of adaptive immunity.
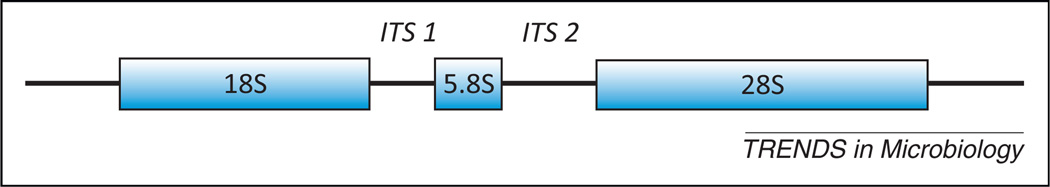
Fungal ribosomal gene locus. The most commonly used genetic region used for culture-independent molecular analysis of fungal diversity. Both clone library/sanger sequencing and pyrosequencing have been used to map and catalogue this region. High-throughput pyrosequencing has been used for amplicon libraries generated through pan-fungal internal transcribed spacer regions (ITS) primers that span the region between the 3′ end of the 18S gene, includes the entire 5S gene, and ends in the 5′ region of the 28S gene [9]. This region has been previously demonstrated to amplify a wide range of medically relevant fungi by PCR and pyrosequencing [–11].
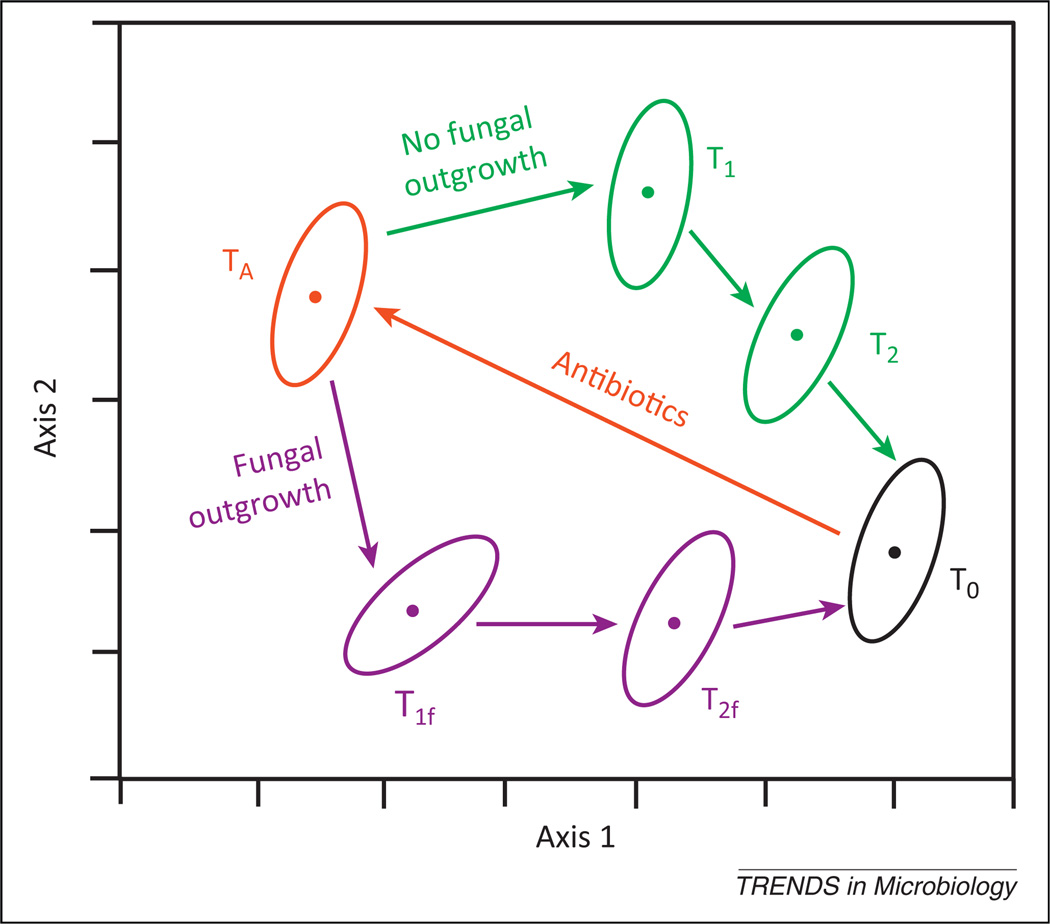
Hypothetical representation of the effect of the presence and absence of a fungal outgrowth that modulates bacterial community reassembly. Reassembly of the bacterial microbiome after antibiotic therapy is an ordered process (TA→T1→T2→T0). Emerging evidence suggests that reassembly may be impacted by the bloom of specific fungal species when colonization resistance is destroyed. This figure provides an illustration of an ecological modeling (ordination) of such a process, in which the bacterial community structure undergoes a different reassembly process (TA→T1f→T2f→T0) in the presence of a fungal bloom. In this example, the differences in bacterial community structure (membership, richness, and evenness) between the TA→T1→T2→T0 path and the TA→T1f→T2f→T0 path could manifest as secondary changes in microbiome-mediated physiologic processes such as immune regulation or metabolism. Abbreviation: T, timepoint.
Similar articles
-
Topographical and physiological differences of the skin mycobiome in health and disease.Virulence. 2017 Apr 3;8(3):324-333. doi: 10.1080/21505594.2016.1249093. Epub 2016 Oct 18. Virulence. 2017. PMID: 27754756 Free PMC article. Review.
-
The vaginal mycobiome: A contemporary perspective on fungi in women's health and diseases.Virulence. 2017 Apr 3;8(3):342-351. doi: 10.1080/21505594.2016.1237332. Epub 2016 Sep 22. Virulence. 2017. PMID: 27657355 Free PMC article. Review.
-
The gut mycobiome of the Human Microbiome Project healthy cohort.Microbiome. 2017 Nov 25;5(1):153. doi: 10.1186/s40168-017-0373-4. Microbiome. 2017. PMID: 29178920 Free PMC article.
-
Review article: fungal microbiota and digestive diseases.Aliment Pharmacol Ther. 2014 Apr;39(8):751-66. doi: 10.1111/apt.12665. Epub 2014 Feb 24. Aliment Pharmacol Ther. 2014. PMID: 24612332 Review.
-
Fungi in the healthy human gastrointestinal tract.Virulence. 2017 Apr 3;8(3):352-358. doi: 10.1080/21505594.2016.1247140. Epub 2016 Oct 13. Virulence. 2017. PMID: 27736307 Free PMC article. Review.
Cited by
-
Analysis of fungal diversity in the feces of Arborophila rufipectus.Front Vet Sci. 2024 Oct 14;11:1430518. doi: 10.3389/fvets.2024.1430518. eCollection 2024. Front Vet Sci. 2024. PMID: 39469585 Free PMC article.
-
Changes in the composition of intestinal fungi and their role in mice with dextran sulfate sodium-induced colitis.Sci Rep. 2015 May 27;5:10416. doi: 10.1038/srep10416. Sci Rep. 2015. PMID: 26013555 Free PMC article.
-
Investigation into In Vitro and In Vivo Caenorhabditis elegans Models to Select Cheese Yeasts as Probiotic Candidates for their Preventive Effects against Salmonella Typhimurium.Microorganisms. 2020 Jun 18;8(6):922. doi: 10.3390/microorganisms8060922. Microorganisms. 2020. PMID: 32570901 Free PMC article.
-
Morphological and physiological changes induced by contact-dependent interaction between Candida albicans and Fusobacterium nucleatum.Sci Rep. 2016 Jun 14;6:27956. doi: 10.1038/srep27956. Sci Rep. 2016. PMID: 27295972 Free PMC article.
-
Dynamics of the Gut Bacteria and Fungi Accompanying Low-Carbohydrate Diet-Induced Weight Loss in Overweight and Obese Adults.Front Nutr. 2022 Feb 11;9:846378. doi: 10.3389/fnut.2022.846378. eCollection 2022. Front Nutr. 2022. PMID: 35223965 Free PMC article.
References
Publication types
MeSH terms
Grants and funding
- U19 AI090871/AI/NIAID NIH HHS/United States
- R01 AI064479/AI/NIAID NIH HHS/United States
- R01HL114447/HL/NHLBI NIH HHS/United States
- U01HL98961/HL/NHLBI NIH HHS/United States
- R01 HL114447/HL/NHLBI NIH HHS/United States
- P30 DK034933/DK/NIDDK NIH HHS/United States
- R01AI072406/AI/NIAID NIH HHS/United States
- U01 HL098961/HL/NHLBI NIH HHS/United States
- R01DE022069/DE/NIDCR NIH HHS/United States
- R21 AI087869/AI/NIAID NIH HHS/United States
- U19AI090871/AI/NIAID NIH HHS/United States
- R01 DE022069/DE/NIDCR NIH HHS/United States
- R01 AI072406/AI/NIAID NIH HHS/United States
- R21 AI083473/AI/NIAID NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
