

A functional deficiency of TERA/VCP/p97 contributes to impaired DNA repair in multiple polyglutamine diseases
- PMID: 23652004
- PMCID: PMC4543262
- DOI: 10.1038/ncomms2828
A functional deficiency of TERA/VCP/p97 contributes to impaired DNA repair in multiple polyglutamine diseases
Abstract
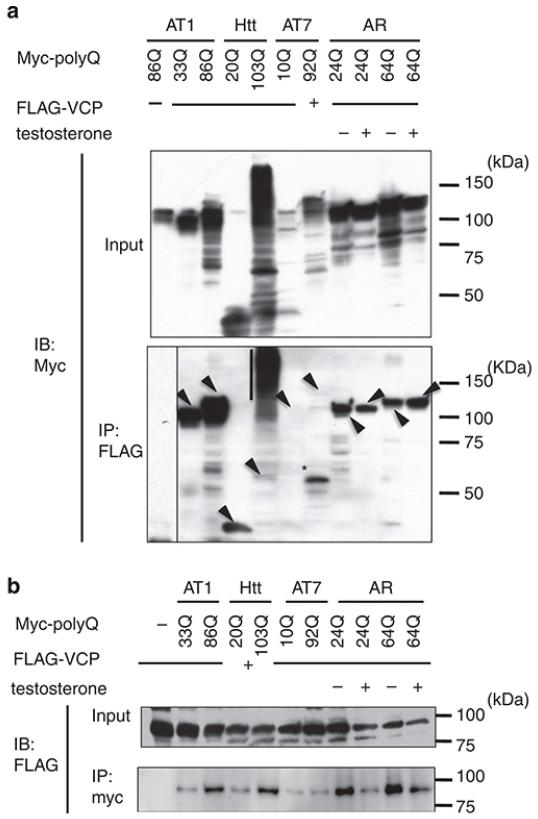
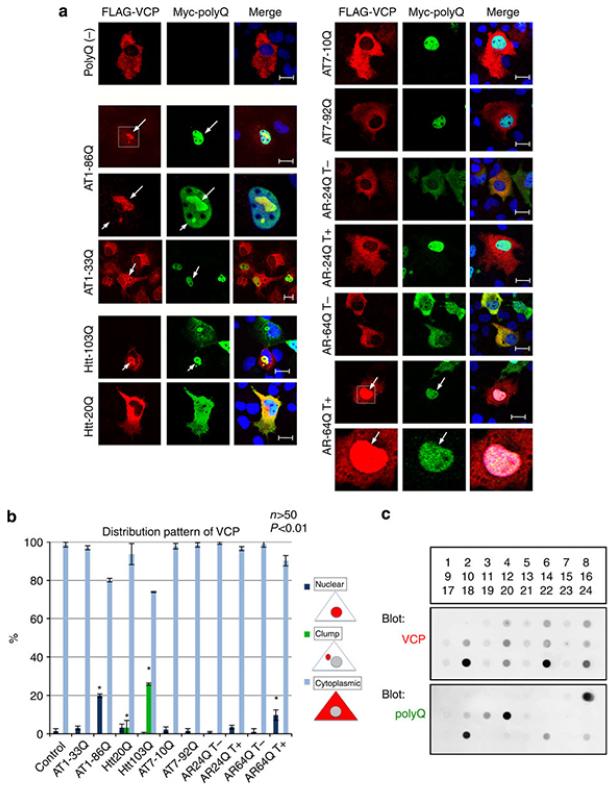
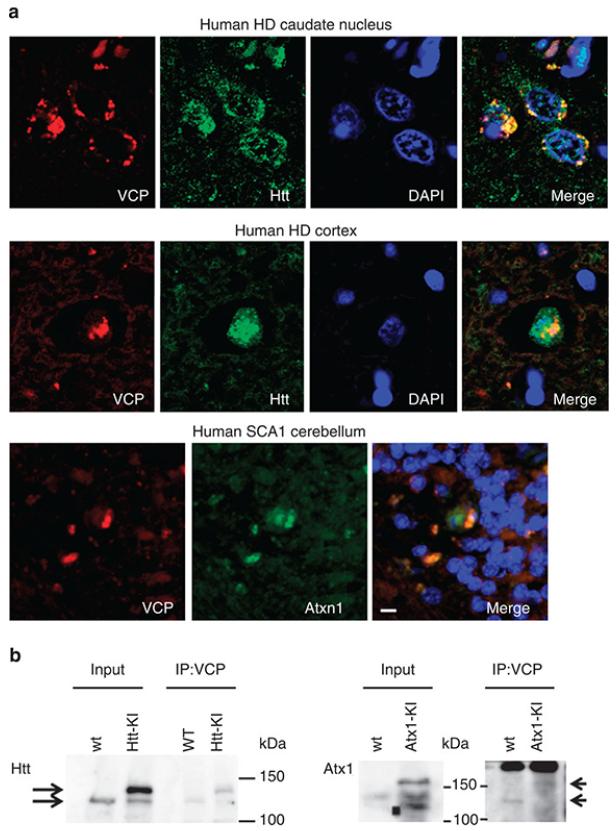
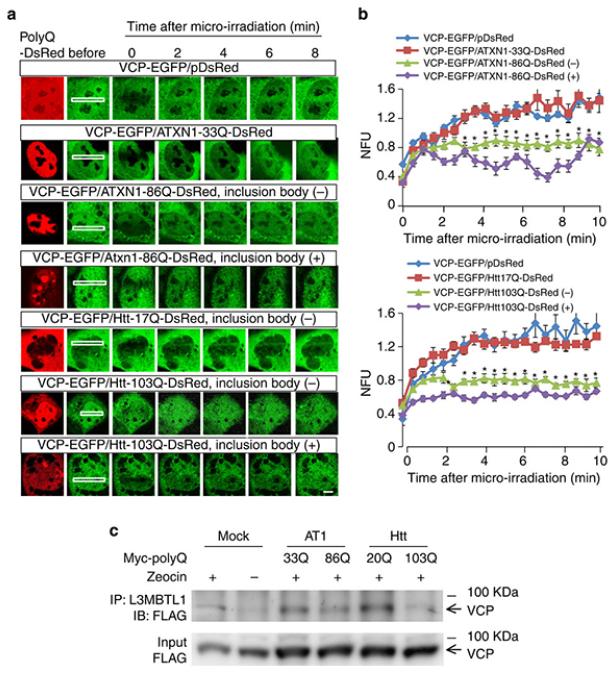
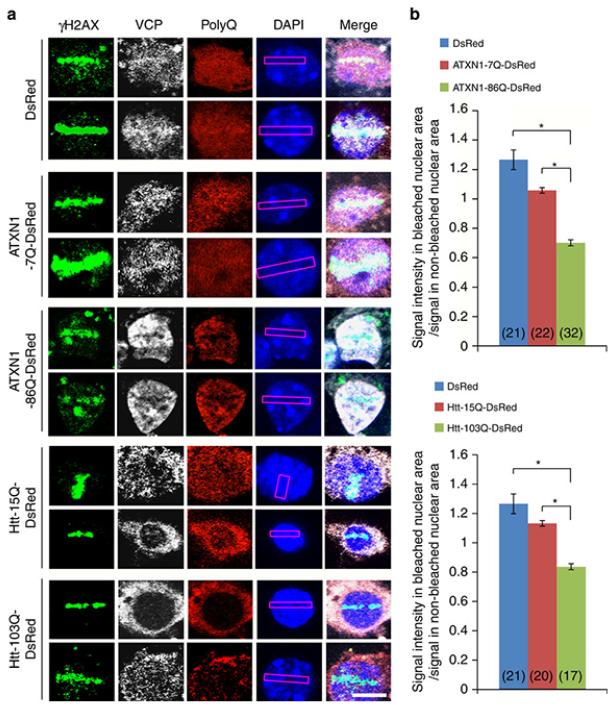
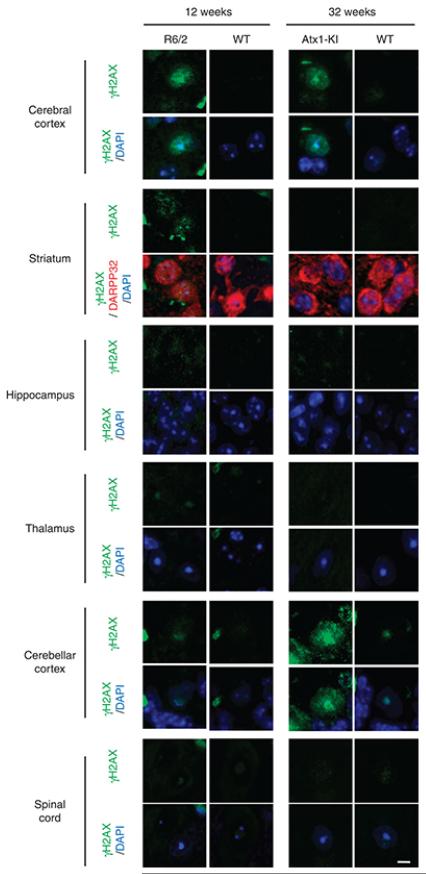
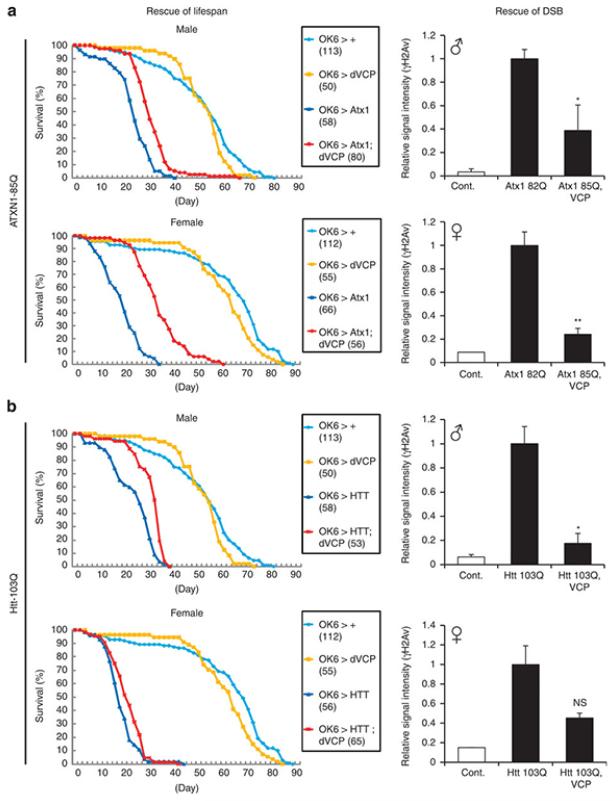
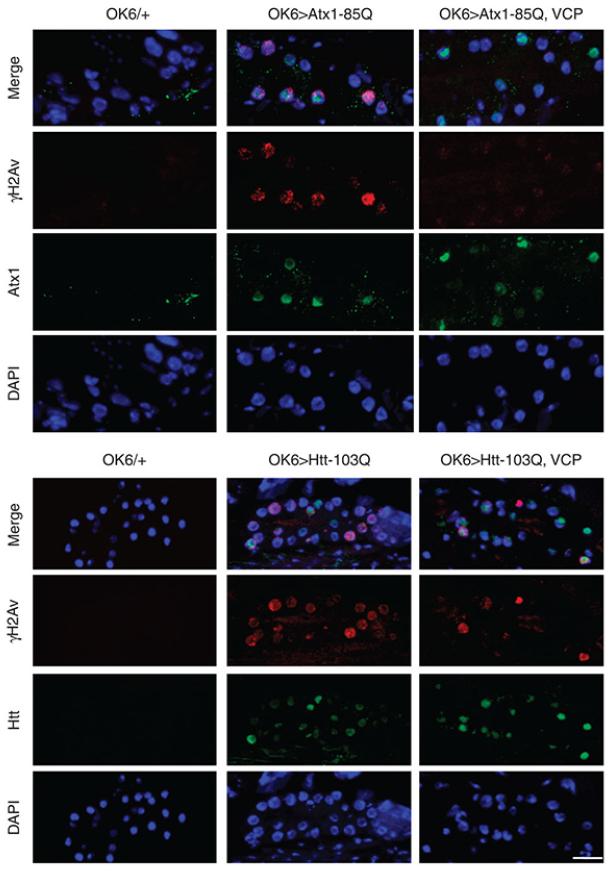
It is hypothesized that a common underlying mechanism links multiple neurodegenerative disorders. Here we show that transitional endoplasmic reticulum ATPase (TERA)/valosin-containing protein (VCP)/p97 directly binds to multiple polyglutamine disease proteins (huntingtin, ataxin-1, ataxin-7 and androgen receptor) via polyglutamine sequence. Although normal and mutant polyglutamine proteins interact with TERA/VCP/p97, only mutant proteins affect dynamism of TERA/VCP/p97. Among multiple functions of TERA/VCP/p97, we reveal that functional defect of TERA/VCP/p97 in DNA double-stranded break repair is critical for the pathology of neurons in which TERA/VCP/p97 is located dominantly in the nucleus in vivo. Mutant polyglutamine proteins impair accumulation of TERA/VCP/p97 and interaction of related double-stranded break repair proteins, finally causing the increase of unrepaired double-stranded break. Consistently, the recovery of lifespan in polyglutamine disease fly models by TERA/VCP/p97 corresponds well to the improvement of double-stranded break in neurons. Taken together, our results provide a novel common pathomechanism in multiple polyglutamine diseases that is mediated by DNA repair function of TERA/VCP/p97.
Figures
Similar articles
-
Valosin-containing protein (VCP/p97) is an activator of wild-type ataxin-3.PLoS One. 2012;7(9):e43563. doi: 10.1371/journal.pone.0043563. Epub 2012 Sep 6. PLoS One. 2012. PMID: 22970133 Free PMC article.
-
Ataxin-3 binds VCP/p97 and regulates retrotranslocation of ERAD substrates.Hum Mol Genet. 2006 Aug 15;15(16):2409-20. doi: 10.1093/hmg/ddl164. Epub 2006 Jul 5. Hum Mol Genet. 2006. PMID: 16822850
-
Functional ATPase activity of p97/valosin-containing protein (VCP) is required for the quality control of endoplasmic reticulum in neuronally differentiated mammalian PC12 cells.J Biol Chem. 2002 Dec 6;277(49):47358-65. doi: 10.1074/jbc.M207783200. Epub 2002 Sep 25. J Biol Chem. 2002. PMID: 12351637
-
Valosin-Containing Protein (VCP)/p97: A Prognostic Biomarker and Therapeutic Target in Cancer.Int J Mol Sci. 2021 Sep 21;22(18):10177. doi: 10.3390/ijms221810177. Int J Mol Sci. 2021. PMID: 34576340 Free PMC article. Review.
-
The VCP/p97 system at a glance: connecting cellular function to disease pathogenesis.J Cell Sci. 2014 Sep 15;127(Pt 18):3877-83. doi: 10.1242/jcs.093831. Epub 2014 Aug 21. J Cell Sci. 2014. PMID: 25146396 Free PMC article. Review.
Cited by
-
PQBP3 prevents senescence by suppressing PSME3-mediated proteasomal Lamin B1 degradation.EMBO J. 2024 Sep;43(18):3968-3999. doi: 10.1038/s44318-024-00192-4. Epub 2024 Aug 5. EMBO J. 2024. PMID: 39103492 Free PMC article.
-
Mitochondrial Abnormalities and Synaptic Damage in Huntington's Disease: a Focus on Defective Mitophagy and Mitochondria-Targeted Therapeutics.Mol Neurobiol. 2021 Dec;58(12):6350-6377. doi: 10.1007/s12035-021-02556-x. Epub 2021 Sep 14. Mol Neurobiol. 2021. PMID: 34519969 Review.
-
Identification of Novel SCIRR69-Interacting Proteins During ER Stress Using SILAC-Immunoprecipitation Quantitative Proteomics Approach.Neuromolecular Med. 2017 Mar;19(1):81-93. doi: 10.1007/s12017-016-8431-9. Epub 2016 Aug 3. Neuromolecular Med. 2017. PMID: 27488499
-
Reduction in PA28αβ activation in HD mouse brain correlates to increased mHTT aggregation in cell models.PLoS One. 2022 Dec 27;17(12):e0278130. doi: 10.1371/journal.pone.0278130. eCollection 2022. PLoS One. 2022. PMID: 36574405 Free PMC article.
-
Meta-analysis of DNA double-strand break response kinetics.Nucleic Acids Res. 2017 Dec 15;45(22):12625-12637. doi: 10.1093/nar/gkx1128. Nucleic Acids Res. 2017. PMID: 29182755 Free PMC article.
References
-
- Watts GD, et al. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet. 2004;36:377–381. - PubMed
-
- Imafuku I, et al. Polar amino acid-rich sequences bind to polyglutamine tracts. Biochem Biophys Res Commun. 1998;253:16–20. - PubMed
-
- Hirabayashi M, et al. VCP/p97 in abnormal protein aggregates, cytoplasmic vacuoles, and cell death, phenotypes relevant to neurodegeneration. Cell Death Differ. 2001;8:977–984. - PubMed
-
- Woodman PG. p97, a protein coping with multiple identities. J Cell Sci. 2003;116:4283–4290. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
