

Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses
- PMID: 23651856
- PMCID: PMC3656320
- DOI: 10.1101/gad.212282.112
Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses
Abstract
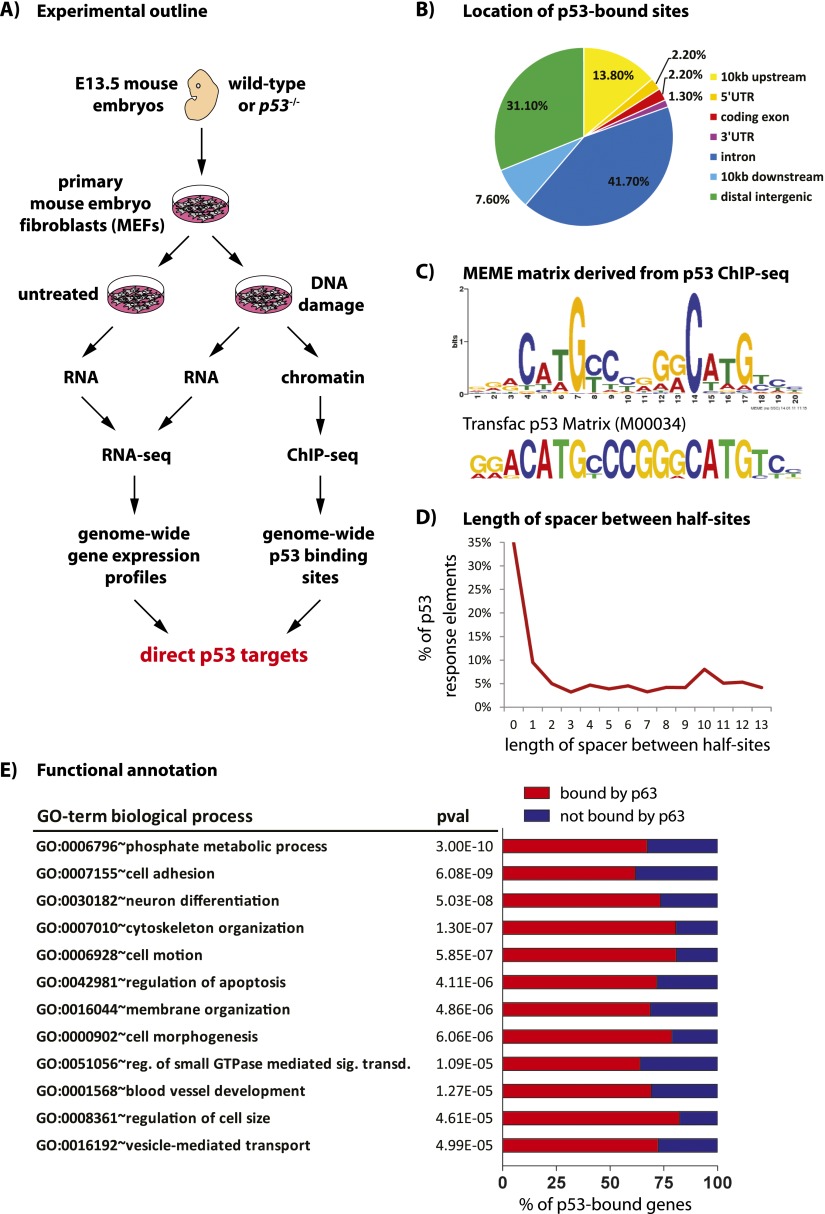
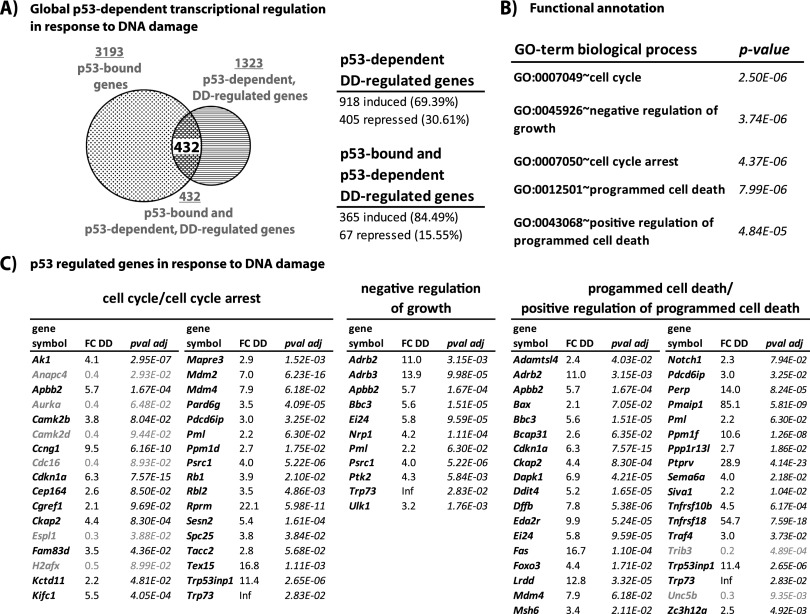
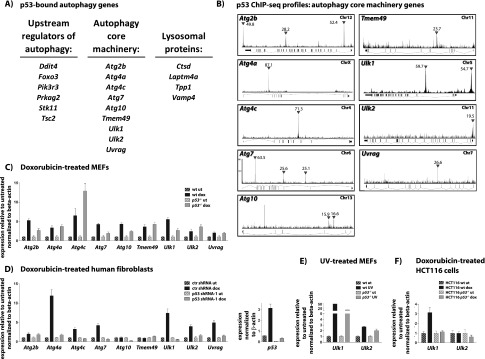
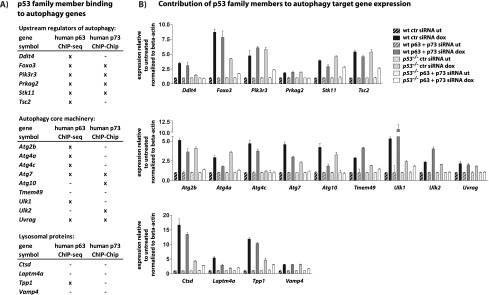
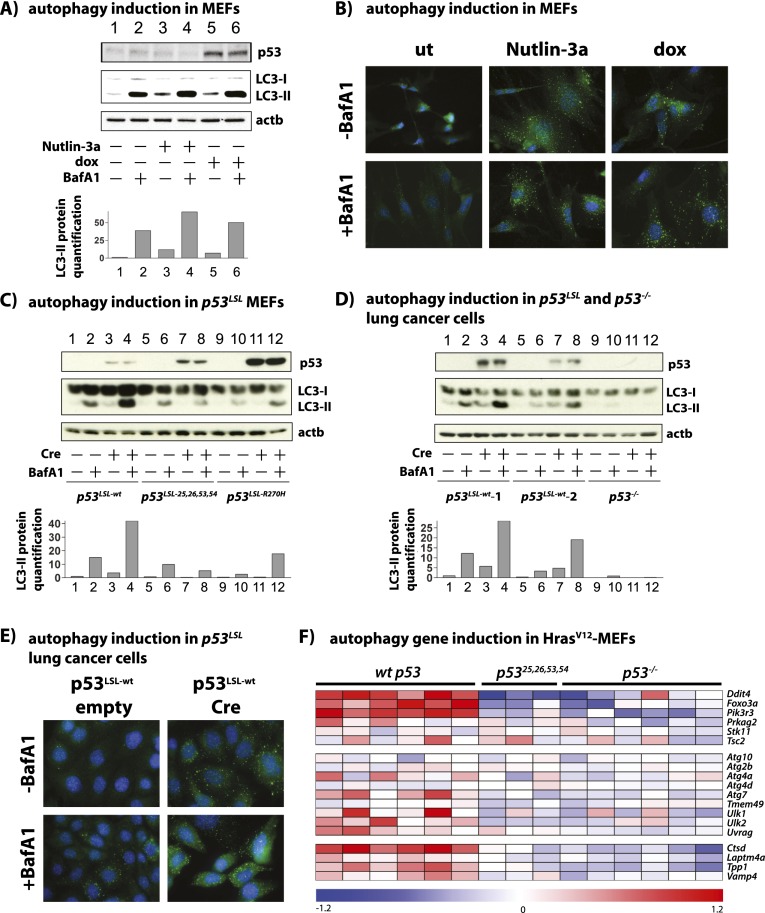
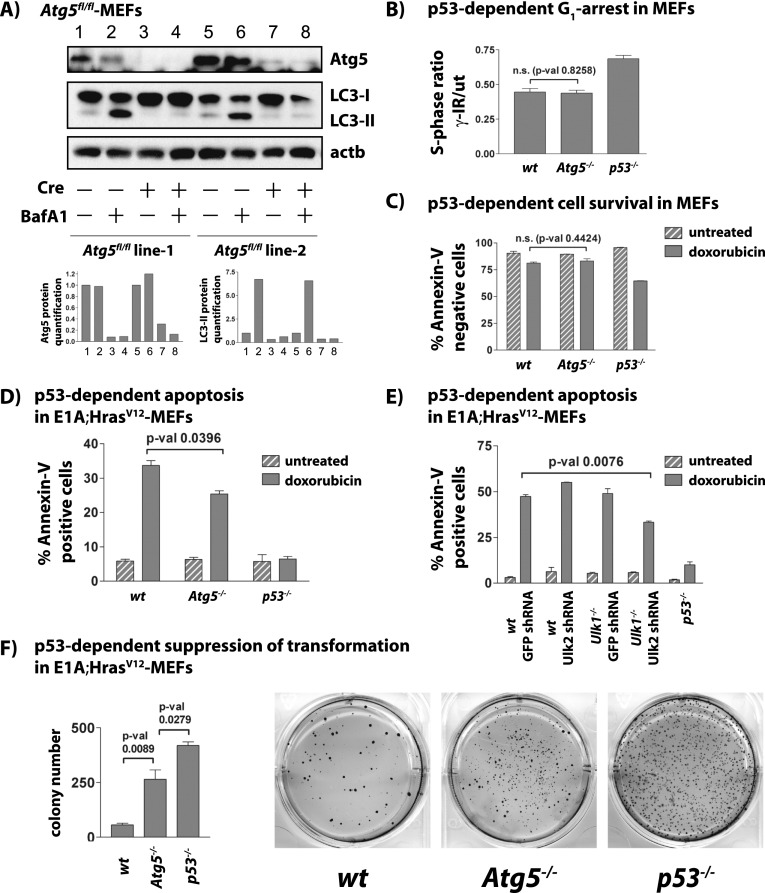
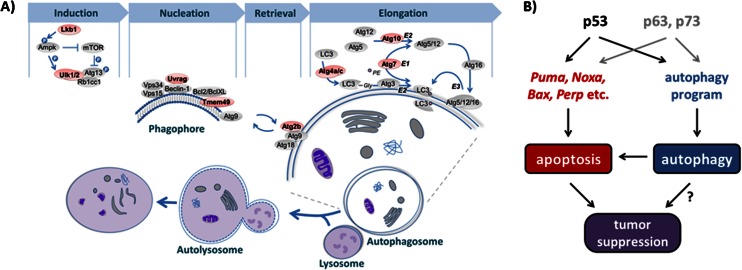
The mechanisms by which the p53 tumor suppressor acts remain incompletely understood. To gain new insights into p53 biology, we used high-throughput sequencing to analyze global p53 transcriptional networks in primary mouse embryo fibroblasts in response to DNA damage. Chromatin immunoprecipitation sequencing reveals 4785 p53-bound sites in the genome located near 3193 genes involved in diverse biological processes. RNA sequencing analysis shows that only a subset of p53-bound genes is transcriptionally regulated, yielding a list of 432 p53-bound and regulated genes. Interestingly, we identify a host of autophagy genes as direct p53 target genes. While the autophagy program is regulated predominantly by p53, the p53 family members p63 and p73 contribute to activation of this autophagy gene network. Induction of autophagy genes in response to p53 activation is associated with enhanced autophagy in diverse settings and depends on p53 transcriptional activity. While p53-induced autophagy does not affect cell cycle arrest in response to DNA damage, it is important for both robust p53-dependent apoptosis triggered by DNA damage and transformation suppression by p53. Together, our data highlight an intimate connection between p53 and autophagy through a vast transcriptional network and indicate that autophagy contributes to p53-dependent apoptosis and cancer suppression.
Keywords: ChIP-seq; RNA-seq; autophagy; p53; tumor suppression.
Figures
Comment in
-
The family that eats together stays together: new p53 family transcriptional targets in autophagy.Genes Dev. 2013 May 1;27(9):971-4. doi: 10.1101/gad.219147.113. Genes Dev. 2013. PMID: 23651851 Free PMC article.
Similar articles
-
The family that eats together stays together: new p53 family transcriptional targets in autophagy.Genes Dev. 2013 May 1;27(9):971-4. doi: 10.1101/gad.219147.113. Genes Dev. 2013. PMID: 23651851 Free PMC article.
-
TRP53 activates a global autophagy program to promote tumor suppression.Autophagy. 2013 Sep;9(9):1440-2. doi: 10.4161/auto.25833. Epub 2013 Jul 29. Autophagy. 2013. PMID: 23899499 Free PMC article.
-
Transcriptional profiling in C. elegans suggests DNA damage dependent apoptosis as an ancient function of the p53 family.BMC Genomics. 2008 Jul 15;9:334. doi: 10.1186/1471-2164-9-334. BMC Genomics. 2008. PMID: 18627611 Free PMC article.
-
p53 and Tumor Suppression: It Takes a Network.Trends Cell Biol. 2021 Apr;31(4):298-310. doi: 10.1016/j.tcb.2020.12.011. Epub 2021 Jan 28. Trends Cell Biol. 2021. PMID: 33518400 Free PMC article. Review.
-
p53 and metabolism: Inside the TIGAR.Cell. 2006 Jul 14;126(1):30-2. doi: 10.1016/j.cell.2006.06.032. Cell. 2006. PMID: 16839873 Review.
Cited by
-
HIF1α Represses Cell Stress Pathways to Allow Proliferation of Hypoxic Fetal Cardiomyocytes.Dev Cell. 2015 Jun 8;33(5):507-21. doi: 10.1016/j.devcel.2015.04.021. Epub 2015 May 28. Dev Cell. 2015. PMID: 26028220 Free PMC article.
-
Identification of novel radiation-induced p53-dependent transcripts extensively regulated during mouse brain development.Biol Open. 2015 Feb 13;4(3):331-44. doi: 10.1242/bio.20149969. Biol Open. 2015. PMID: 25681390 Free PMC article.
-
Differential Protein Distribution between the Nucleus and Mitochondria: Implications in Aging.Front Genet. 2016 Sep 16;7:162. doi: 10.3389/fgene.2016.00162. eCollection 2016. Front Genet. 2016. PMID: 27695477 Free PMC article. Review.
-
Lipid Droplets in Cancer: Guardians of Fat in a Stressful World.Molecules. 2018 Aug 3;23(8):1941. doi: 10.3390/molecules23081941. Molecules. 2018. PMID: 30081476 Free PMC article. Review.
-
p53 restoration in small cell lung cancer identifies a latent cyclophilin-dependent necrosis mechanism.Nat Commun. 2023 Jul 21;14(1):4403. doi: 10.1038/s41467-023-40161-9. Nat Commun. 2023. PMID: 37479684 Free PMC article.
References
-
- Bailey TL, Elkan C 1994. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol 2: 28–36 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous