

Stepwise protein folding at near amino acid resolution by hydrogen exchange and mass spectrometry
- PMID: 23603271
- PMCID: PMC3651421
- DOI: 10.1073/pnas.1305887110
Stepwise protein folding at near amino acid resolution by hydrogen exchange and mass spectrometry
Abstract
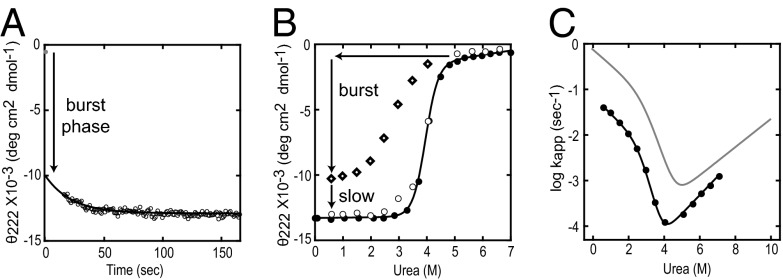
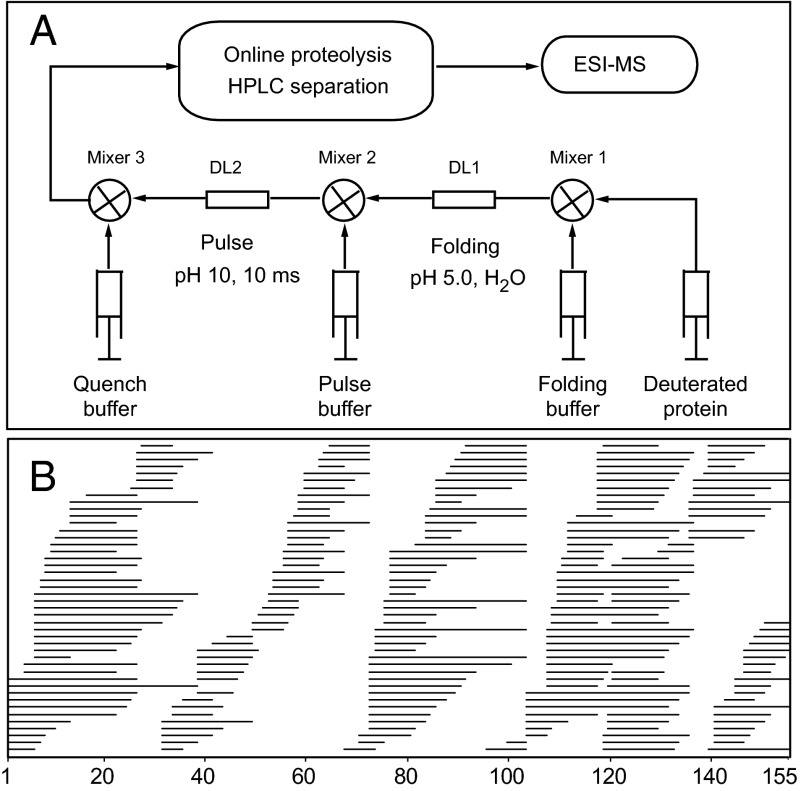
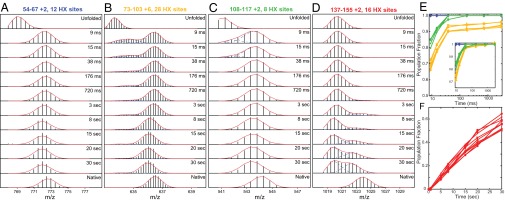
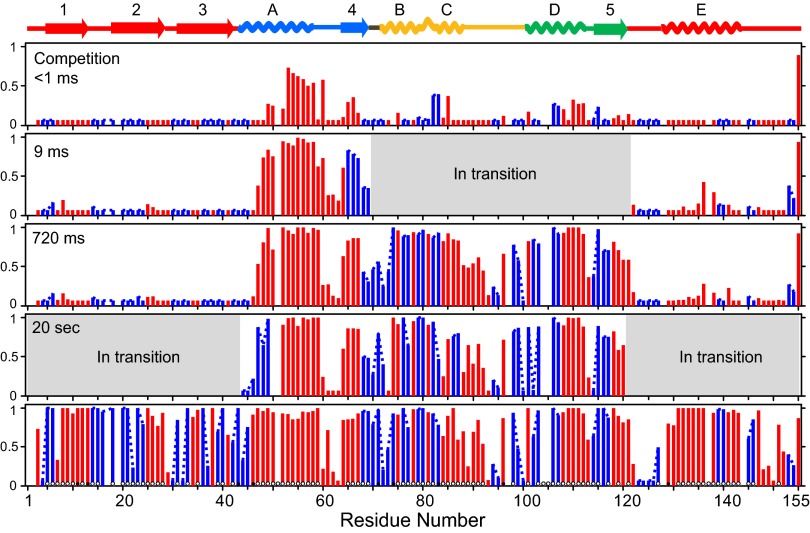
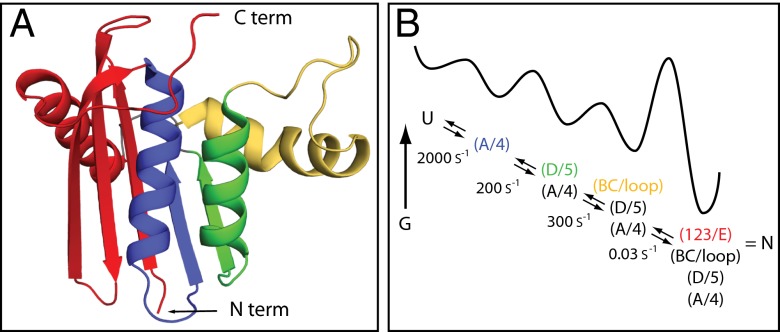
The kinetic folding of ribonuclease H was studied by hydrogen exchange (HX) pulse labeling with analysis by an advanced fragment separation mass spectrometry technology. The results show that folding proceeds through distinct intermediates in a stepwise pathway that sequentially incorporates cooperative native-like structural elements to build the native protein. Each step is seen as a concerted transition of one or more segments from an HX-unprotected to an HX-protected state. Deconvolution of the data to near amino acid resolution shows that each step corresponds to the folding of a secondary structural element of the native protein, termed a "foldon." Each folded segment is retained through subsequent steps of foldon addition, revealing a stepwise buildup of the native structure via a single dominant pathway. Analysis of the pertinent literature suggests that this model is consistent with experimental results for many proteins and some current theoretical results. Two biophysical principles appear to dictate this behavior. The principle of cooperativity determines the central role of native-like foldon units. An interaction principle termed "sequential stabilization" based on native-like interfoldon interactions orders the pathway.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Protein Folding-How and Why: By Hydrogen Exchange, Fragment Separation, and Mass Spectrometry.Annu Rev Biophys. 2016 Jul 5;45:135-52. doi: 10.1146/annurev-biophys-062215-011121. Epub 2016 Apr 27. Annu Rev Biophys. 2016. PMID: 27145881 Free PMC article. Review.
-
An amino acid code for protein folding.Proc Natl Acad Sci U S A. 2001 Jan 2;98(1):105-12. doi: 10.1073/pnas.98.1.105. Proc Natl Acad Sci U S A. 2001. PMID: 11136249 Free PMC article.
-
Structural analysis of kinetic folding intermediates for a TIM barrel protein, indole-3-glycerol phosphate synthase, by hydrogen exchange mass spectrometry and Gō model simulation.J Mol Biol. 2007 Nov 23;374(2):528-46. doi: 10.1016/j.jmb.2007.09.024. Epub 2007 Sep 14. J Mol Biol. 2007. PMID: 17942114 Free PMC article.
-
Cooperative folding near the downhill limit determined with amino acid resolution by hydrogen exchange.Proc Natl Acad Sci U S A. 2016 Apr 26;113(17):4747-52. doi: 10.1073/pnas.1522500113. Epub 2016 Apr 13. Proc Natl Acad Sci U S A. 2016. PMID: 27078098 Free PMC article.
-
Hydrogen exchange methods to study protein folding.Methods. 2004 Sep;34(1):51-64. doi: 10.1016/j.ymeth.2004.03.005. Methods. 2004. PMID: 15283915 Review.
Cited by
-
High-resolution epitope mapping by HX MS reveals the pathogenic mechanism and a possible therapy for autoimmune TTP syndrome.Proc Natl Acad Sci U S A. 2015 Aug 4;112(31):9620-5. doi: 10.1073/pnas.1512561112. Epub 2015 Jul 22. Proc Natl Acad Sci U S A. 2015. PMID: 26203127 Free PMC article.
-
Protein Folding-How and Why: By Hydrogen Exchange, Fragment Separation, and Mass Spectrometry.Annu Rev Biophys. 2016 Jul 5;45:135-52. doi: 10.1146/annurev-biophys-062215-011121. Epub 2016 Apr 27. Annu Rev Biophys. 2016. PMID: 27145881 Free PMC article. Review.
-
A conserved folding nucleus sculpts the free energy landscape of bacterial and archaeal orthologs from a divergent TIM barrel family.Proc Natl Acad Sci U S A. 2021 Apr 27;118(17):e2019571118. doi: 10.1073/pnas.2019571118. Proc Natl Acad Sci U S A. 2021. PMID: 33875592 Free PMC article.
-
Protein Folding Structures: Formation of Folding Structures Based on Probability Theory.ACS Omega. 2016 Dec 22;1(6):1355-1366. doi: 10.1021/acsomega.6b00206. eCollection 2016 Dec 31. ACS Omega. 2016. PMID: 31457201 Free PMC article.
-
Revealing what gets buried first in protein folding.Proc Natl Acad Sci U S A. 2013 Oct 15;110(42):16704-5. doi: 10.1073/pnas.1316158110. Epub 2013 Oct 4. Proc Natl Acad Sci U S A. 2013. PMID: 24096579 Free PMC article. No abstract available.
References
-
- Chamberlain AK, Handel TM, Marqusee S. Detection of rare partially folded molecules in equilibrium with the native conformation of RNaseH. Nat Struct Biol. 1996;3(9):782–787. - PubMed
-
- Raschke TM, Marqusee S. The kinetic folding intermediate of ribonuclease H resembles the acid molten globule and partially unfolded molecules detected under native conditions. Nat Struct Biol. 1997;4(4):298–304. - PubMed
-
- Raschke TM, Kho J, Marqusee S. Confirmation of the hierarchical folding of RNase H: A protein engineering study. Nat Struct Biol. 1999;6(9):825–831. - PubMed
-
- Cecconi C, Shank EA, Bustamante C, Marqusee S. Direct observation of the three-state folding of a single protein molecule. Science. 2005;309(5743):2057–2060. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
