

Insights into congenital stationary night blindness based on the structure of G90D rhodopsin
- PMID: 23579341
- PMCID: PMC3674435
- DOI: 10.1038/embor.2013.44
Insights into congenital stationary night blindness based on the structure of G90D rhodopsin
Abstract
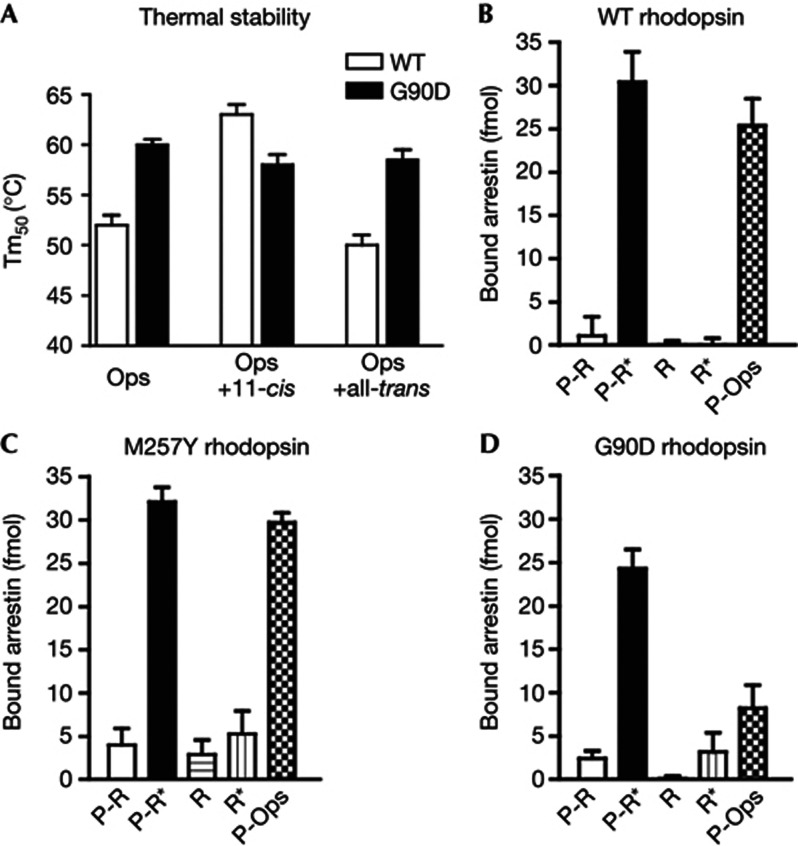
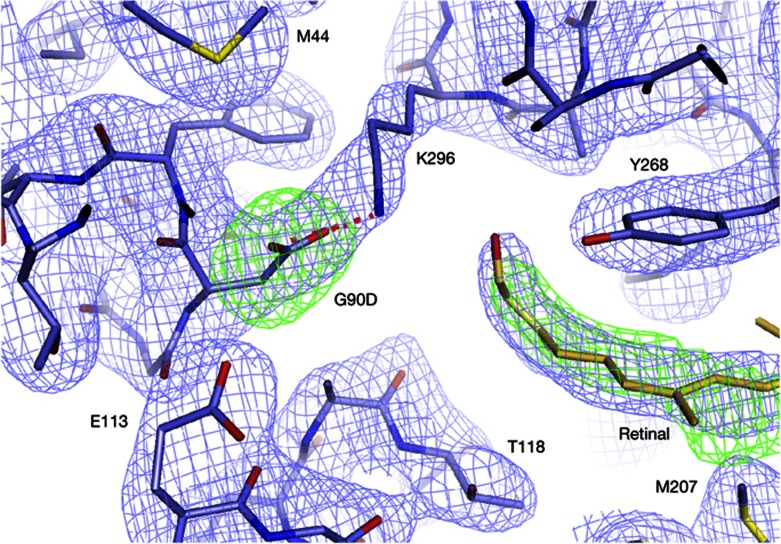
We present active-state structures of the G protein-coupled receptor (GPCRs) rhodopsin carrying the disease-causing mutation G90D. Mutations of G90 cause either retinitis pigmentosa (RP) or congenital stationary night blindness (CSNB), a milder, non-progressive form of RP. Our analysis shows that the CSNB-causing G90D mutation introduces a salt bridge with K296. The mutant thus interferes with the E113Q-K296 activation switch and the covalent binding of the inverse agonist 11-cis-retinal, two interactions that are crucial for the deactivation of rhodopsin. Other mutations, including G90V causing RP, cannot promote similar interactions. We discuss our findings in context of a model in which CSNB is caused by constitutive activation of the visual signalling cascade.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures



Similar articles
-
Structural role of the T94I rhodopsin mutation in congenital stationary night blindness.EMBO Rep. 2016 Oct;17(10):1431-1440. doi: 10.15252/embr.201642671. Epub 2016 Jul 25. EMBO Rep. 2016. PMID: 27458239 Free PMC article.
-
Structural, energetic, and mechanical perturbations in rhodopsin mutant that causes congenital stationary night blindness.J Biol Chem. 2012 Jun 22;287(26):21826-35. doi: 10.1074/jbc.M112.340182. Epub 2012 May 1. J Biol Chem. 2012. PMID: 22549882 Free PMC article.
-
Molecular mechanisms of disease for mutations at Gly-90 in rhodopsin.J Biol Chem. 2011 Nov 18;286(46):39993-40001. doi: 10.1074/jbc.M110.201517. Epub 2011 Sep 22. J Biol Chem. 2011. PMID: 21940625 Free PMC article.
-
Structural aspects of rod opsin and their implication in genetic diseases.Pflugers Arch. 2021 Sep;473(9):1339-1359. doi: 10.1007/s00424-021-02546-x. Epub 2021 Mar 16. Pflugers Arch. 2021. PMID: 33728518 Review.
-
1 rhodopsin mutations in congenital night blindness.Adv Exp Med Biol. 2010;664:263-72. doi: 10.1007/978-1-4419-1399-9_30. Adv Exp Med Biol. 2010. PMID: 20238025 Review.
Cited by
-
Structural role of the T94I rhodopsin mutation in congenital stationary night blindness.EMBO Rep. 2016 Oct;17(10):1431-1440. doi: 10.15252/embr.201642671. Epub 2016 Jul 25. EMBO Rep. 2016. PMID: 27458239 Free PMC article.
-
Constitutively active rhodopsin mutants causing night blindness are effectively phosphorylated by GRKs but differ in arrestin-1 binding.Cell Signal. 2013 Nov;25(11):2155-62. doi: 10.1016/j.cellsig.2013.07.009. Epub 2013 Jul 17. Cell Signal. 2013. PMID: 23872075 Free PMC article.
-
Arrestins in apoptosis.Handb Exp Pharmacol. 2014;219:309-39. doi: 10.1007/978-3-642-41199-1_16. Handb Exp Pharmacol. 2014. PMID: 24292837 Free PMC article.
-
Modeling of Olfactory Receptors.Methods Mol Biol. 2023;2627:183-193. doi: 10.1007/978-1-0716-2974-1_10. Methods Mol Biol. 2023. PMID: 36959448
-
Retinal degeneration in mice expressing the constitutively active G90D rhodopsin mutant.Hum Mol Genet. 2020 Apr 15;29(6):881-891. doi: 10.1093/hmg/ddaa008. Hum Mol Genet. 2020. PMID: 31960909 Free PMC article.
References
-
- Neidhardt J, Barthelmes D, Farahmand F, Fleischhauer JC, Berger W (2006) Different amino acid substitutions at the same position in rhodopsin lead to distinct phenotypes. Invest Ophthalmol Vis Sci 47: 1630–1635 - PubMed
-
- Rao VR, Cohen GB, Oprian DD (1994) Rhodopsin mutation G90D and a molecular mechanism for congenital night blindness. Nature 367: 639–642 - PubMed
-
- Zvyaga TA, Fahmy K, Siebert F, Sakmar TP (1996) Characterization of the mutant visual pigment responsible for congenital night blindness: a biochemical and Fourier-transform infrared spectroscopy study. Biochemistry 35: 7536–7545 - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
