

Polo-like kinase 1 inhibitors, mitotic stress and the tumor suppressor p53
- PMID: 23574746
- PMCID: PMC3674062
- DOI: 10.4161/cc.24573
Polo-like kinase 1 inhibitors, mitotic stress and the tumor suppressor p53
Abstract
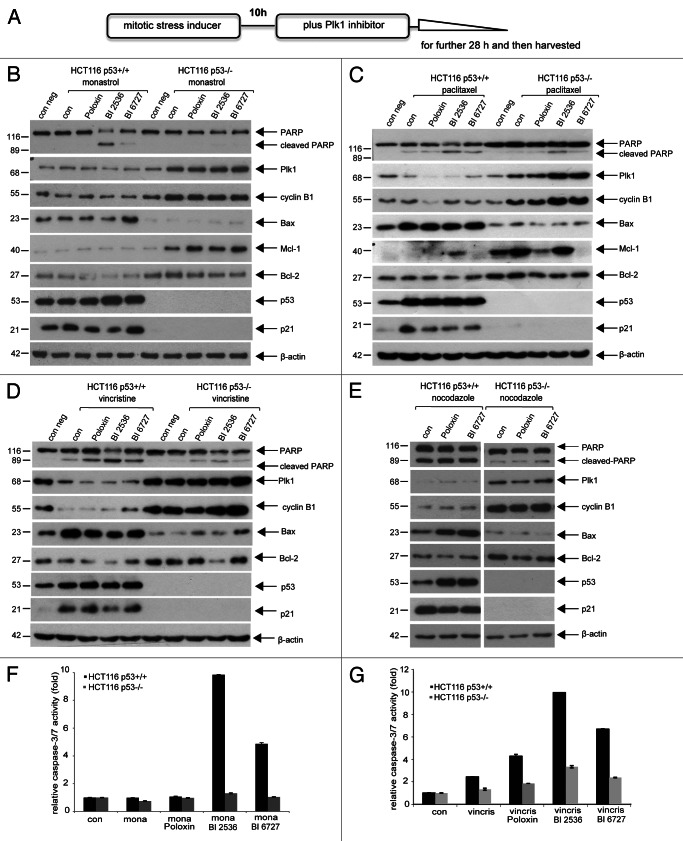
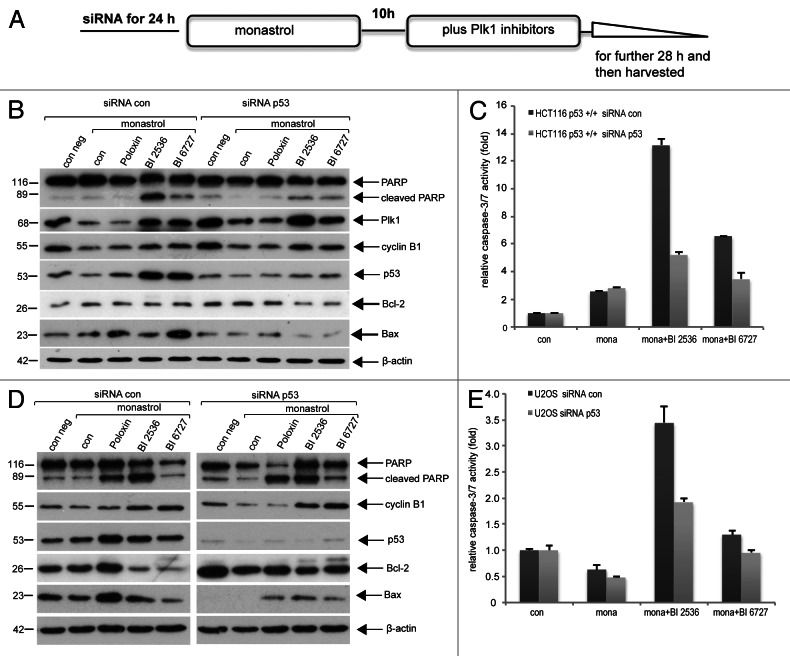
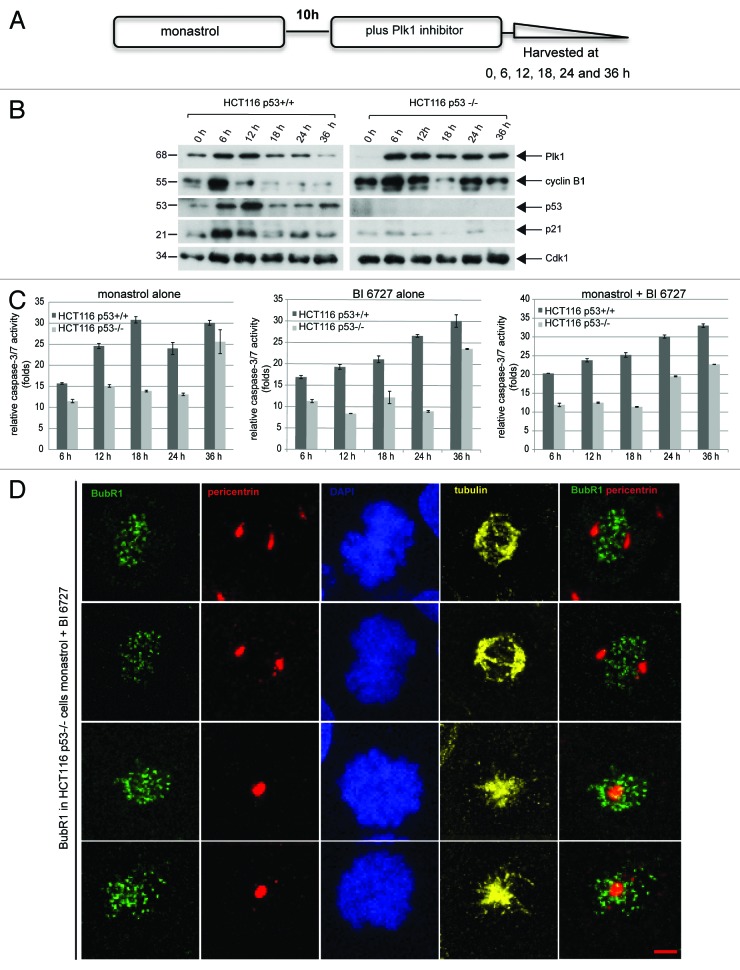
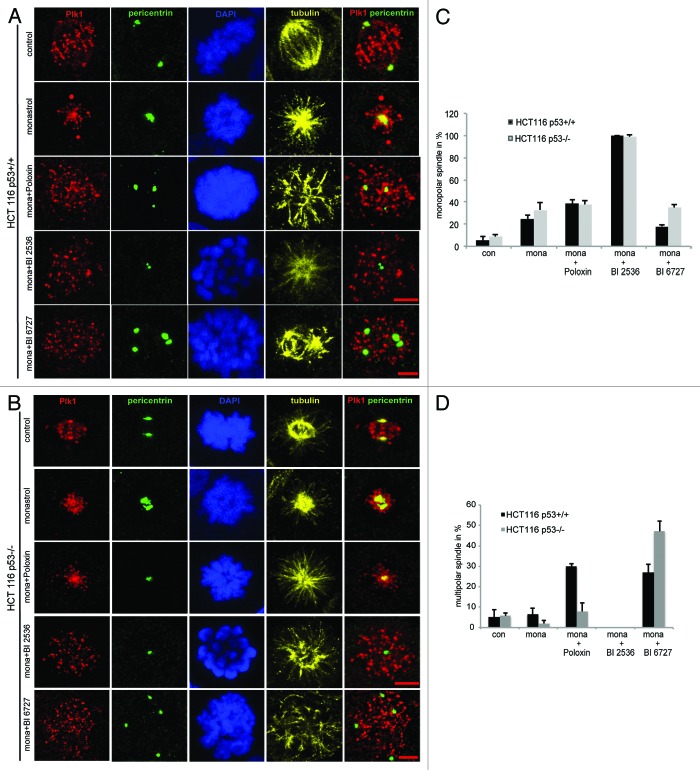
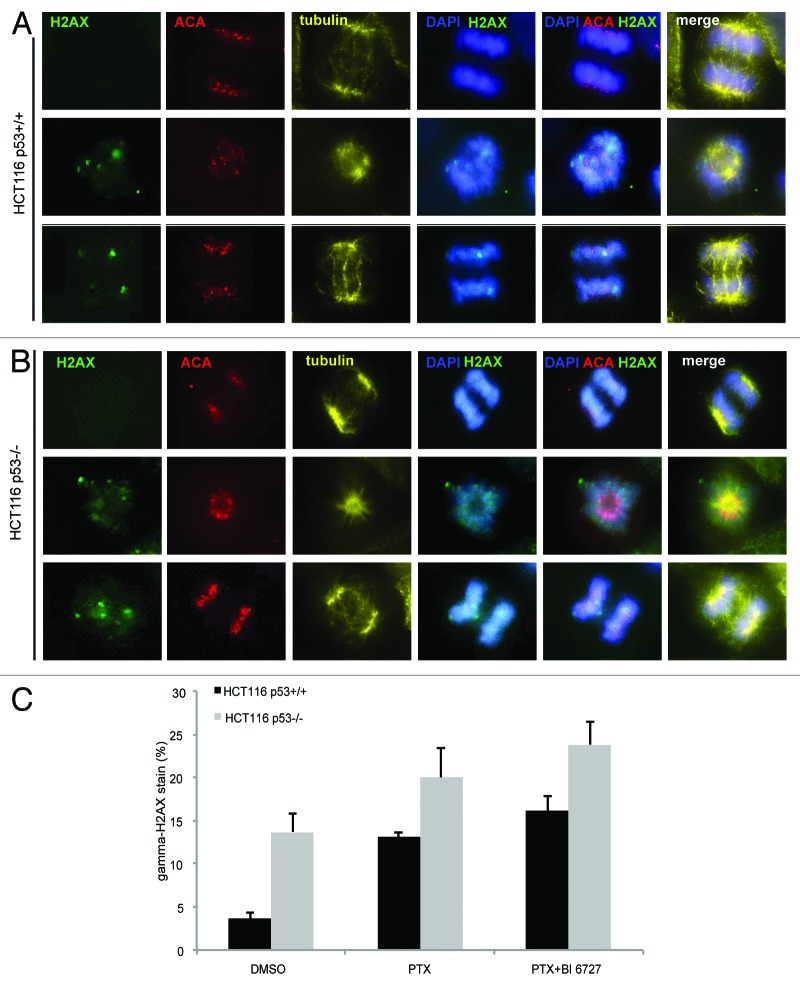
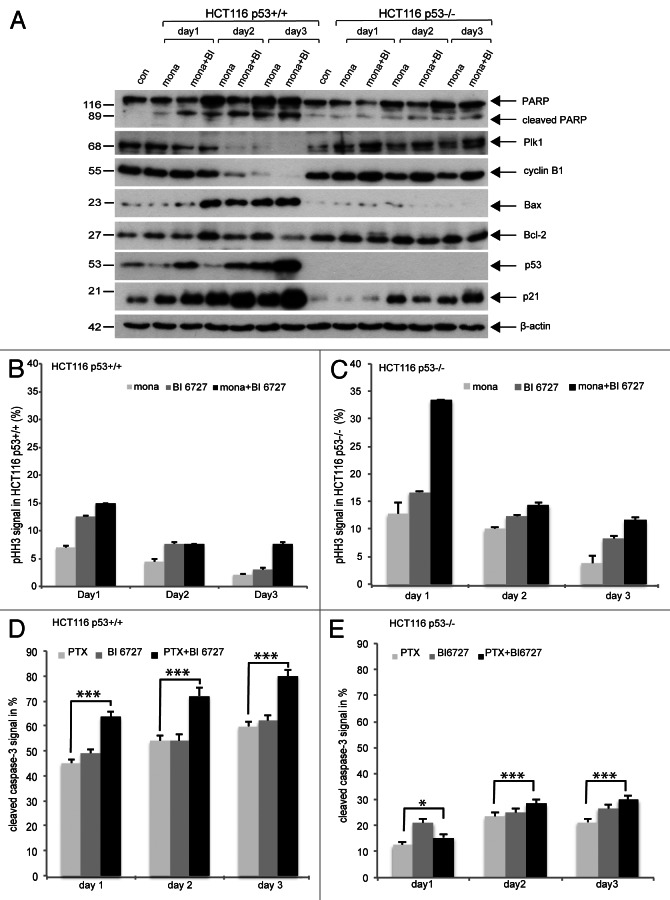
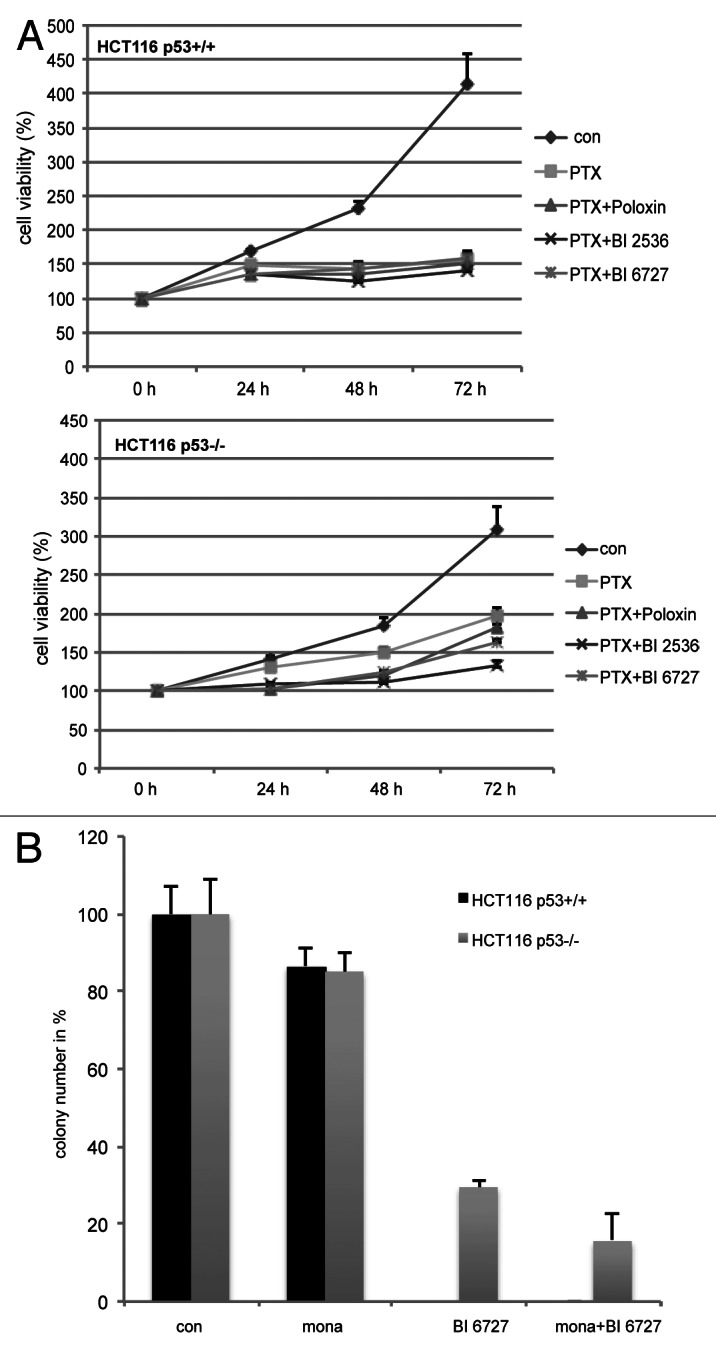
Polo-like kinase 1 has been established as one of the most attractive targets for molecular cancer therapy. In fact, multiple small-molecule inhibitors targeting this kinase have been developed and intensively investigated. Recently, it has been reported that the cytotoxicity induced by Plk1 inhibition is elevated in cancer cells with inactive p53, leading to the hypothesis that inactive p53 is a predictive marker for the response of Plk1 inhibition. In our previous study based on different cancer cell lines, we showed that cancer cells with wild type p53 were more sensitive to Plk1 inhibition by inducing more apoptosis, compared with cancer cells depleted of p53. In the present work, we further demonstrate that in the presence of mitotic stress induced by different agents, Plk1 inhibitors strongly induced apoptosis in HCT116 p53(+/+) cells, whereas HCT116 p53(-/-) cells arrested in mitosis with less apoptosis. Depletion of p53 in HCT116 p53(+/+) or U2OS cells reduced the induction of apoptosis. Moreover, the surviving HCT116 p53(-/-) cells showed DNA damage and a strong capability of colony formation. Plk1 inhibition in combination with other anti-mitotic agents inhibited proliferation of tumor cells more strongly than Plk1 inhibition alone. Taken together, the data underscore that functional p53 strengthens the efficacy of Plk1 inhibition alone or in combination by strongly activating cell death signaling pathways. Further studies are required to investigate if the long-term outcomes of losing p53, such as low differential grade of tumor cells or defective DNA damage checkpoint, are responsible for the cytotoxicity of Plk1 inhibition.
Keywords: BI 2536; BI 6727; Poloxin; monastrol; p53.
Figures
Similar articles
-
p53 is not directly relevant to the response of Polo-like kinase 1 inhibitors.Cell Cycle. 2012 Feb 1;11(3):543-53. doi: 10.4161/cc.11.3.19076. Epub 2012 Feb 1. Cell Cycle. 2012. PMID: 22262171
-
Differential Cellular Effects of Plk1 Inhibitors Targeting the ATP-binding Domain or Polo-box Domain.J Cell Physiol. 2015 Dec;230(12):3057-67. doi: 10.1002/jcp.25042. J Cell Physiol. 2015. PMID: 25975351
-
Impact of Polo-like kinase 1 inhibitors on human adipose tissue-derived mesenchymal stem cells.Oncotarget. 2016 Dec 20;7(51):84271-84285. doi: 10.18632/oncotarget.12482. Oncotarget. 2016. PMID: 27713178 Free PMC article.
-
Polo-like Kinase 1 as an emerging drug target: structure, function and therapeutic implications.J Drug Target. 2021 Feb;29(2):168-184. doi: 10.1080/1061186X.2020.1818760. Epub 2020 Sep 14. J Drug Target. 2021. PMID: 32886539 Review.
-
Non-mitotic functions of polo-like kinases in cancer cells.Biochim Biophys Acta Rev Cancer. 2021 Jan;1875(1):188467. doi: 10.1016/j.bbcan.2020.188467. Epub 2020 Nov 7. Biochim Biophys Acta Rev Cancer. 2021. PMID: 33171265 Review.
Cited by
-
B-cell lymphoma 6 promotes proliferation and survival of trophoblastic cells.Cell Cycle. 2016;15(6):827-39. doi: 10.1080/15384101.2016.1149273. Cell Cycle. 2016. PMID: 27029530 Free PMC article.
-
Target Analysis and Mechanism of Podophyllotoxin in the Treatment of Triple-Negative Breast Cancer.Front Pharmacol. 2020 Aug 7;11:1211. doi: 10.3389/fphar.2020.01211. eCollection 2020. Front Pharmacol. 2020. PMID: 32848800 Free PMC article.
-
Adenovirus replaces mitotic checkpoint controls.J Virol. 2015 May;89(9):5083-96. doi: 10.1128/JVI.00213-15. Epub 2015 Feb 18. J Virol. 2015. PMID: 25694601 Free PMC article.
-
Loss of p21Cip1/CDKN1A renders cancer cells susceptible to Polo-like kinase 1 inhibition.Oncotarget. 2015 Mar 30;6(9):6611-26. doi: 10.18632/oncotarget.2844. Oncotarget. 2015. PMID: 25483104 Free PMC article.
-
Functional analysis of phosphorylation of the mitotic centromere-associated kinesin by Aurora B kinase in human tumor cells.Cell Cycle. 2015;14(23):3755-67. doi: 10.1080/15384101.2015.1068481. Epub 2015 Jul 6. Cell Cycle. 2015. PMID: 26148251 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous