

S-nitrosylation of ARH is required for LDL uptake by the LDL receptor
- PMID: 23564733
- PMCID: PMC3646456
- DOI: 10.1194/jlr.M033167
S-nitrosylation of ARH is required for LDL uptake by the LDL receptor
Abstract
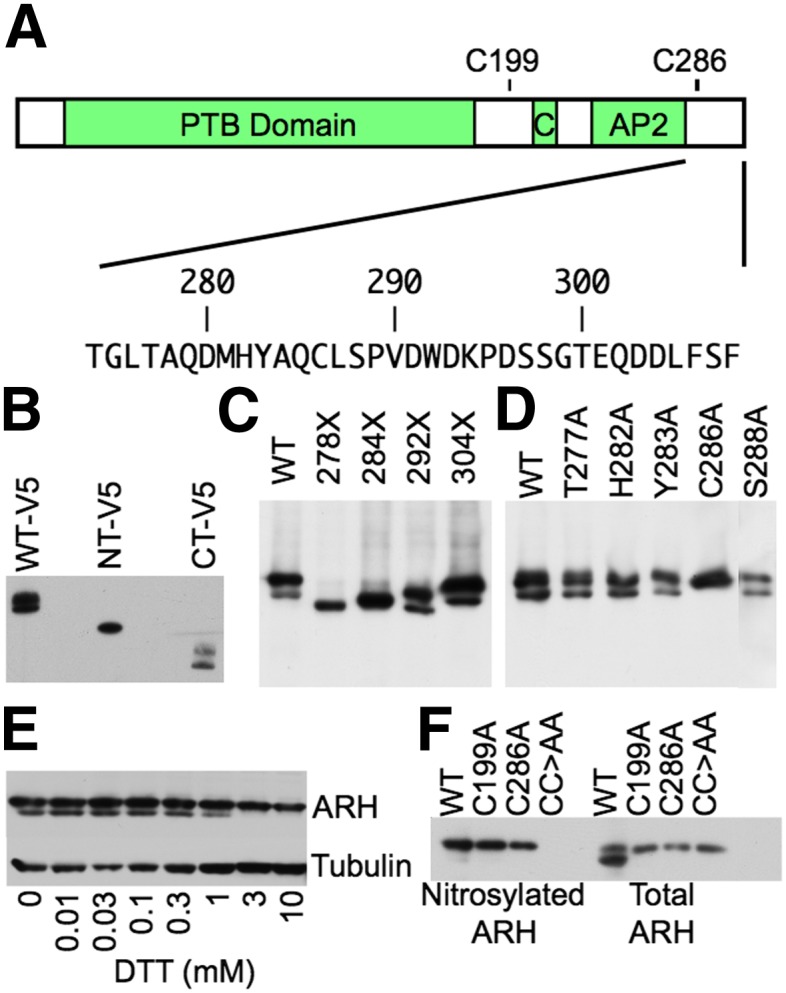
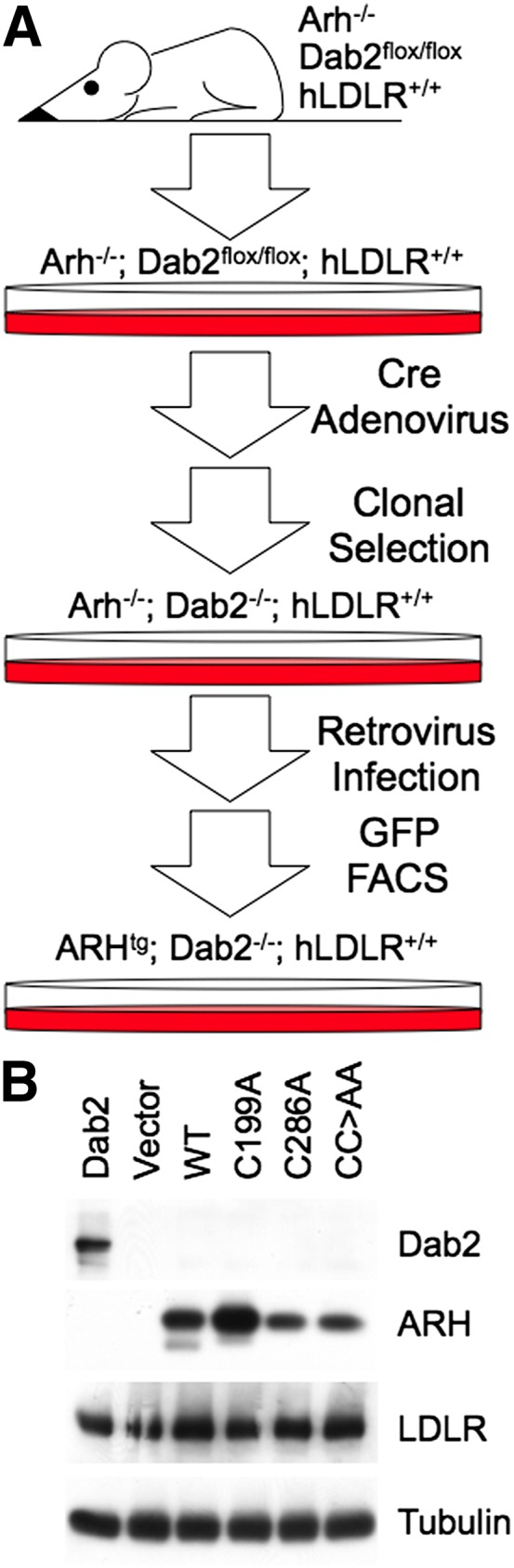
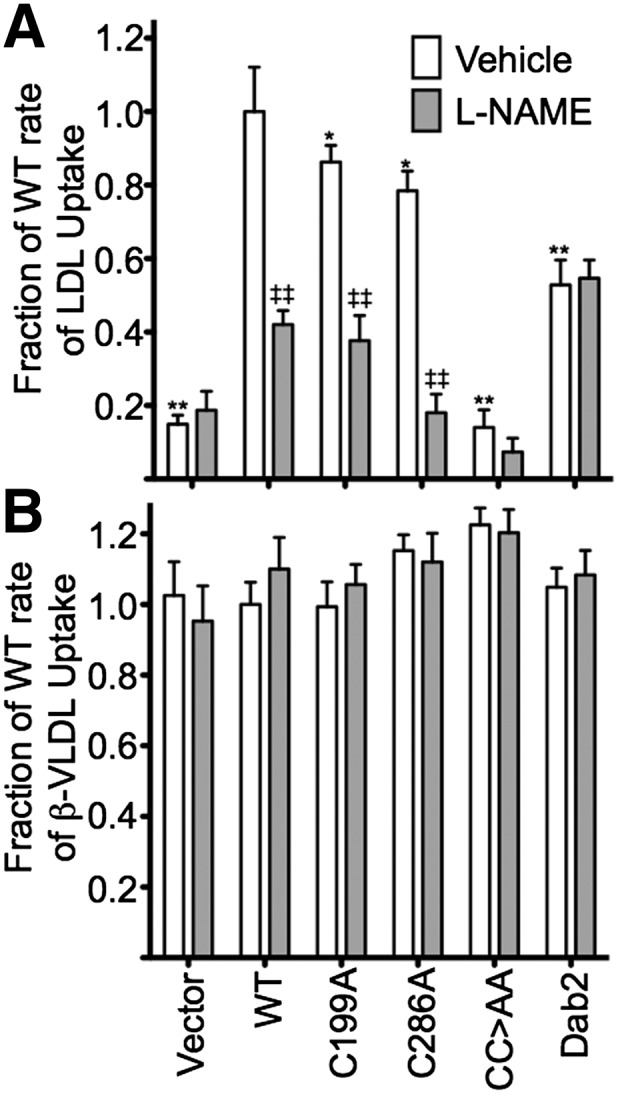
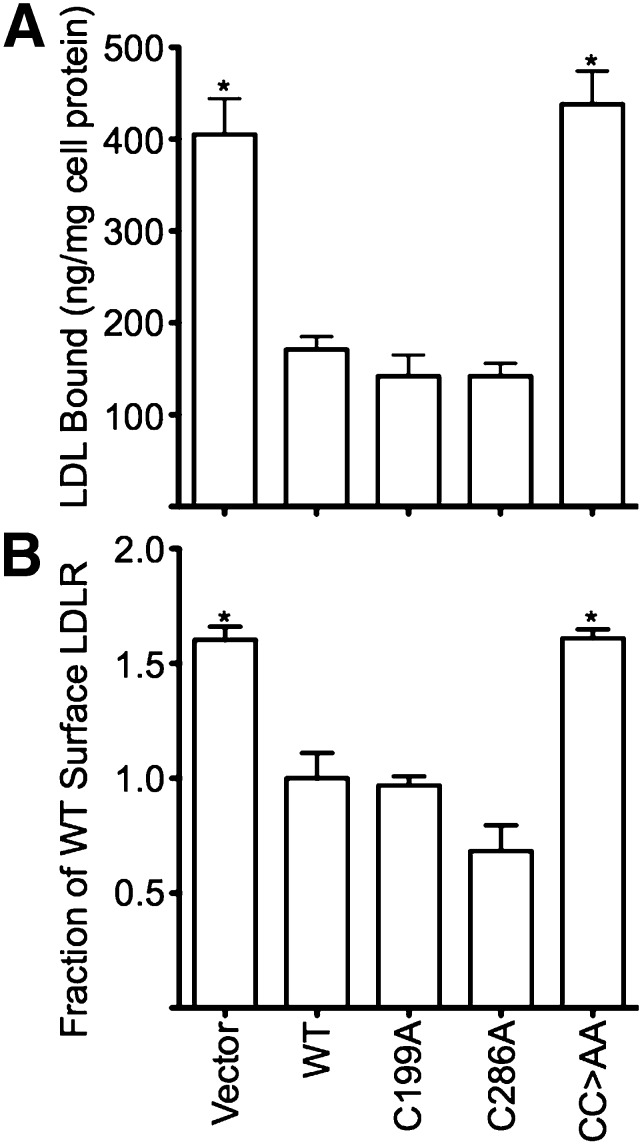
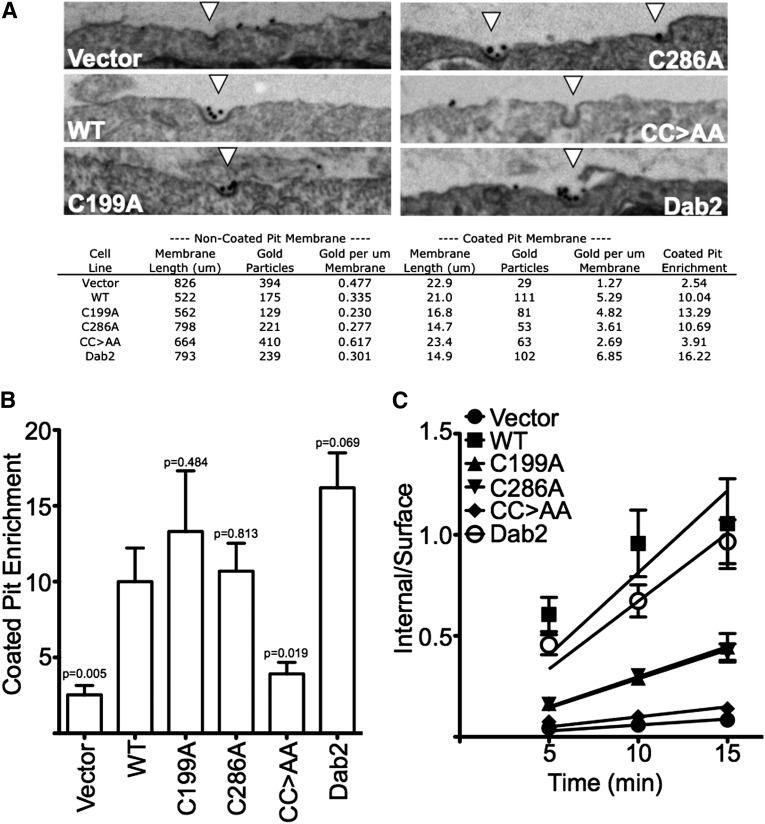
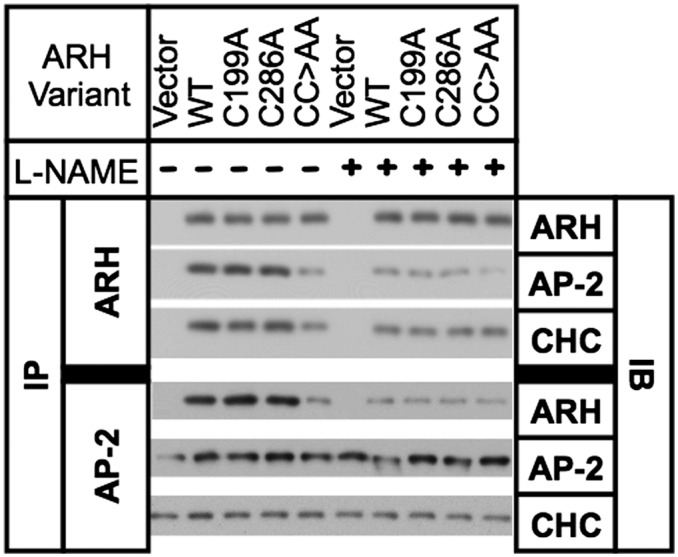
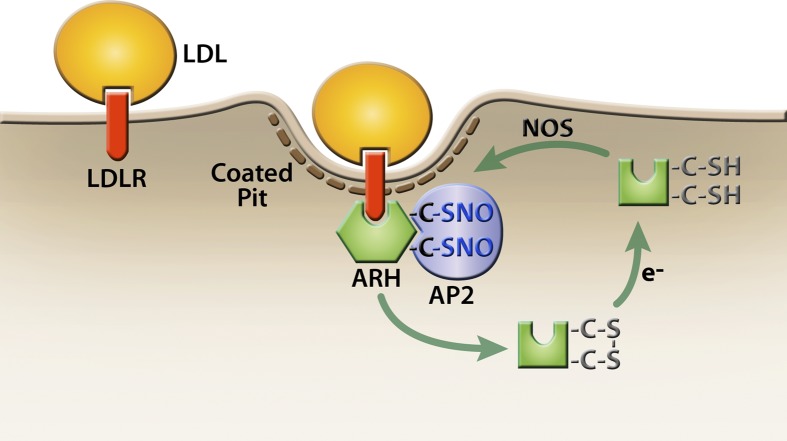
The LDL receptor (LDLR) relies upon endocytic adaptor proteins for internalization of lipoproteins. The results of this study show that the LDLR adaptor autosomal recessive hypercholesterolemia protein (ARH) requires nitric oxide to support LDL uptake. Nitric oxide nitrosylates ARH at C199 and C286, and these posttranslational modifications are necessary for association of ARH with the adaptor protein 2 (AP-2) component of clathrin-coated pits. In the absence of nitrosylation, ARH is unable to target LDL-LDLR complexes to coated pits, resulting in poor LDL uptake. The role of nitric oxide on LDLR function is specific for ARH because inhibition of nitric oxide synthase activity impairs ARH-supported LDL uptake but has no effect on other LDLR-dependent lipoprotein uptake processes, including VLDL remnant uptake and dab2-supported LDL uptake. These findings suggest that cells that depend upon ARH for LDL uptake can control which lipoproteins are internalized by their LDLRs through changes in nitric oxide.
Keywords: autosomal recessive hypercholesterolemia protein; low density lipoprotein receptor; nitric oxide.
Figures
Similar articles
-
The adaptor protein Dab2 sorts LDL receptors into coated pits independently of AP-2 and ARH.J Cell Sci. 2006 Oct 15;119(Pt 20):4235-46. doi: 10.1242/jcs.03217. Epub 2006 Sep 19. J Cell Sci. 2006. PMID: 16984970
-
The modular adaptor protein autosomal recessive hypercholesterolemia (ARH) promotes low density lipoprotein receptor clustering into clathrin-coated pits.J Biol Chem. 2005 Dec 9;280(49):40996-1004. doi: 10.1074/jbc.M509394200. Epub 2005 Sep 22. J Biol Chem. 2005. PMID: 16179341
-
The clathrin adaptor proteins ARH, Dab2, and numb play distinct roles in Niemann-Pick C1-Like 1 versus low density lipoprotein receptor-mediated cholesterol uptake.J Biol Chem. 2014 Nov 28;289(48):33689-700. doi: 10.1074/jbc.M114.593764. Epub 2014 Oct 20. J Biol Chem. 2014. PMID: 25331956 Free PMC article.
-
Molecular mechanisms of autosomal recessive hypercholesterolemia.Curr Opin Lipidol. 2003 Apr;14(2):121-7. doi: 10.1097/00041433-200304000-00002. Curr Opin Lipidol. 2003. PMID: 12642779 Review.
-
Autosomal recessive hypercholesterolemia.Semin Vasc Med. 2004 Aug;4(3):241-8. doi: 10.1055/s-2004-861491. Semin Vasc Med. 2004. PMID: 15630633 Review.
Cited by
-
Endocytosis in the adaptation to cellular stress.Cell Stress. 2020 Aug 18;4(10):230-247. doi: 10.15698/cst2020.10.232. Cell Stress. 2020. PMID: 33024932 Free PMC article. Review.
-
Relationship between Brain Metabolic Disorders and Cognitive Impairment: LDL Receptor Defect.Int J Mol Sci. 2022 Jul 29;23(15):8384. doi: 10.3390/ijms23158384. Int J Mol Sci. 2022. PMID: 35955522 Free PMC article. Review.
-
A multienzyme S-nitrosylation cascade regulates cholesterol homeostasis.Cell Rep. 2022 Oct 25;41(4):111538. doi: 10.1016/j.celrep.2022.111538. Cell Rep. 2022. PMID: 36288700 Free PMC article.
-
Endocytic adaptors Arh and Dab2 control homeostasis of circulatory cholesterol.J Lipid Res. 2016 May;57(5):809-17. doi: 10.1194/jlr.M063065. Epub 2016 Mar 22. J Lipid Res. 2016. PMID: 27005486 Free PMC article.
-
Targeting protein modifications in metabolic diseases: molecular mechanisms and targeted therapies.Signal Transduct Target Ther. 2023 May 27;8(1):220. doi: 10.1038/s41392-023-01439-y. Signal Transduct Target Ther. 2023. PMID: 37244925 Free PMC article. Review.
References
-
- James R. W., Martin B., Pometta D., Fruchart J. C., Duriez P., Puchois P., Farriaux J. P., Tacquet A., Demant T., Clegg R. J., et al. 1989. Apolipoprotein B metabolism in homozygous familial hypercholesterolemia. J. Lipid Res. 30: 159–169 - PubMed
-
- Chen W. J., Goldstein J. L., Brown M. S. 1990. NPXY, a sequence often found in cytoplasmic tails, is required for coated pit-mediated internalization of the low density lipoprotein receptor. J. Biol. Chem. 265: 3116–3123 - PubMed
-
- Anderson R. G., Goldstein J. L., Brown M. S. 1977. A mutation that impairs the ability of lipoprotein receptors to localise in coated pits on the cell surface of human fibroblasts. Nature. 270: 695–699 - PubMed
-
- He G., Gupta S., Yi M., Michaely P., Hobbs H. H., Cohen J. C. 2002. ARH is a modular adaptor protein that interacts with the LDL receptor, clathrin, and AP-2. J. Biol. Chem. 277: 44044–44049 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
